Poorly differentiated synovial sarcoma of the vagina: a case report and a clinical literature review
L Minig1, G Farnetano3, M Peiretti1, G Roviglione1, V Zanagnolo1, G Pelosi2 and F Landoni1
20141
220141
L Minig. Email: lucas.minig@ieo.it
Abstract
Synovial sarcomas (SS) account for 5–10% of soft-tissue sarcomas and typically arise in the para-articular regions of adolescents and young adults. Nonetheless, SS can occasionally occur in other regions of the body. Here, we present a first clinical literature report of a patient with an SS arising from the vaginal wall. A 40-year-old patient who presented a necrotic polypoid lesion, measuring 50 mm and extending from the external urethral meatus to the middle part of the anterior vaginal wall. The biopsy showed a poorly differentiated SS with abundant necrosis and a SYT-SSX1 mutation. A staging CT scan was negative for distant metastases. The patient, prior to the radical surgery, received neoadjuvant chemotherapy (ifosfamide and epirubicin) for three cycles. She underwent post-operative external radiotherapy and brachytherapy (50 Gy) due to close margins (<1 mm) in the pathologic specimen. She relapsed 11 and 16 months later with lung metastases, which, both times, were successfully removed by surgical resection. At 24 months from diagnosis, the patient is alive without further evidence of disease. In summary, in the presence of unfavourable prognostic factors, neoadjuvant chemotherapy could be the primary approach to reduce the tumour size and the risk of distant micro-metastases allowing a less aggressive radical surgery if the tumour is located in a non-extremity site. Hence, a multidisciplinary approach, if not influencing overall survival and disease-free survival, may improve the quality of life. In fact, in our patient we obtained a complete clinical control in the pelvis, avoiding pelvic exenteration with neoadjuvant chemotherapy.
Background
Synovial sarcomas account for 5–10% of soft-tissue sarcomas, which typically arise in the para-articular regions of adolescents and young adults. Nonetheless, SS rarely occur in the mediastinum, head and neck region, heart, lung and retroperitoneum. Here, we present a clinical literature report of a patient with a synovial sarcoma arising from the vaginal wall. The immunohistochemical, molecular and ultrastructural details of this case have been previously reported [1].
Case presentation
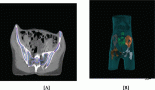
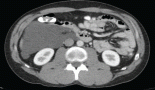
A 40-year-old patient was referred to the European Institute of Oncology (Milan, Italy) with a diagnosis, from elsewhere, of a high-grade vaginal leiomyosarcoma. She was severely anaemic due to abnormal vaginal bleeding, and pelvic examination revealed a necrotic, friable and bluish polypoid lesion, measuring 50 mm longitudinally and extending from the external urethral meatus to the middle part of the anterior vaginal wall. A staging CT scan was negative for distant metastases.
Histopathological review of the original sections revealed a poorly differentiated synovial sarcoma, and the patient was treated with neoadjuvant chemotherapy, including high-dose ifosfamide (9 g/m2) and epirubicin (90 mg/m2), q 21 days, for three cycles, obtaining a partial clinical response (tumour size by clinical examination: 1 cm). She then underwent wide local excision of the anterior vaginal wall, with preservation of the external urethral meatus.
The pathology report confirmed the diagnosis of a poorly differentiated synovial sarcoma and the patient received post-operative external radiotherapy and brachytherapy (50 Gy) for close margins (<1 mm).
Eleven months later the patient experienced three metastases to the right lung, which were treated with wedge resection. Five months later, three other metastases were found on the contralateral lung and were successfully removed by surgical resection.
At 25 months from diagnosis, the patient is alive without further evidence of pulmonary or other metastases or local recurrence.
Pathological and molecular findings
Microscopic examination of this case revealed a high-grade, biphasic, poorly differentiated synovial sarcoma with abundant necrosis and mitoses (more than 10/10 high-power fields). The immunohistochemical findings were consistent with the diagnosis of a poorly differentiated synovial sarcoma, arising from the vaginal wall.
The rearrangement of the SYT gene, characteristic of SS, was detected confirming the diagnosis of synovial sarcoma. A reverse transcriptase-polymerase chain reaction assay revealed a SYT-SSX1 fusion gene product of 158 bp.
Discussion
Synovial sarcomas constitute approximately 5–10% of all soft-tissue sarcomas [2]. Although 90% of cases are diagnosed in the extremities and occur before the age of 50 years, they may arise in a wide variety of organs, including head and neck area, mediastinum, heart, lung, abdominal wall, mesentery, retroperitoneum and peritoneal cavity [3]. In the female genital tract, the occurrence of synovial sarcomas is extremely rare, and only isolated case reports have been described in the fallopian tube, [4] the vulva [5–7] and the vagina [8].
The chance of a synovial sarcoma occurring in other body locations apart from the extremities is supported by the hypothesis that they do not arise from synovial tissue. In fact, it has been suggested that they originate from unknown stem cells that are capable of differentiating into mesenchymal and/or epithelial structures (an hypothesis that still needs to be confirmed) [9]. Biphasic synovial sarcomas contain both epithelial cells arranged in glandular structures and spindle cells, whereas monophasic types are entirely composed of spindle cells. On the other hand, the poorly differentiated type, as the one reported here, often contain small round cells resembling those seen in Ewing's primitive neuroectodermal tumours. However, only immunohistological, ultrastructural and cytogenetic features allow correct diagnosis.
Other types of synovial sarcoma include leiomyosarcoma (which account for the most common histology in any single tumour series of the vagina), the spindle cell variant of squamous cell carcinoma and the rare mixed tumour of the vagina, either benign or malignant.
Synovial sarcomas are characterized by the presence a specific genetic translocation, the SYT-SSX translocation t(X;18) (p11.2;q11.2), which represents a specific diagnostic marker, being present in 95% of all SS cases. Its presence, as the only cytogenetic abnormality, suggests that it is the primary cause of synovial sarcoma.
There are several subtypes, numbered SYT-SSX 1, 2 and 4 [10,11], and these genetic subtypes are correlated with histological subtypes and clinical behaviour. Biphasic synovial sarcomas are associated with the presence of the SYT-SSX1 mutation, while monophasic synovial sarcomas are correlated with the mutation SYT-SSX2. There are data that indicate that a particular subtype of translocation has prognostic significance. In two studies, the SYT-SSX 2 translocation was primarily associated with the monophasic histotype and a better outlook for survival five years after diagnosis, compared with the SYT-SSX 1 translocation, which was primarily associated with biphasic disease and a worse outlook for five-year survival [10,11].
An essential first step in the successful treatment of synovial sarcoma is adequate surgical resection with tumour-free surgical margins [12–14].
However, in cases with unfavourable localization such as the head and neck, retroperitoneum, chest and female genital tract, chemotherapy and, perhaps, pre-operative radiation therapy should be initially given, with the aim of reducing the tumour volume, thus facilitating surgical resection. Our patient received primary chemotherapy with epirubicin and ifosfamide, with good partial remission. Other active regimens can include vincristine, actinomycin D and cyclophosphamide (VAC).
It is interesting to point out that the combination of doxorubicin and ifosfamide induced a more significant responses compared to VAC regimens, as described in two other studies [15,16].
After surgery, our patient received 50 Gy of radiotherapy, as the tumour margins were close (<1 mm) in the pathologic specimen. Given sarcoma's relative rarity, the precise doses of radiotherapy to achieve local control in microscopic and gross residual disease categories are not well known. However, Raney et al [17] usually propose to use a dose of 55 Gy for microscopic or recurrent disease after surgical removal, and somewhat higher, about 60 Gy, in the presence of visible residual tumour.
The natural course of the disease is such that many patients will experience recurrence of the primary tumour and/or metastatic disease. The reported five-year overall survival rates range from 38% to 76% [18,19] and from 20% to 63% in ten years [19,20] . As synovial sarcomas can grow slowly, the ten or 20-year survival rates are more accurate. In fact Paulino [21] found that approximately 26% of distant relapses occurred after five years from the initial diagnosis and 2% were diagnosed 15 years after the initial diagnosis. Metastatic disease occurs in almost 50% of patients. The most common site of metastasis is the lungs (74–81%), lymph nodes (3–23%) and bone (10–20%) [22-24].
The unfavourable prognostic factors, described in several studies, include a tumour size more than 5 cm, tumour localization in a non-extremity site, primary treatment outside a referral centre, a patient age of more than 25 years, the presence of tumour necrosis areas and bone invasion [13,25] and the presence of a particular SYT-SSX translocation (SYT-SSX1) [10]. However, the strongest prognostic factor associated with local recurrence, metastases and tumour-related death in a multivariate analysis [25] is the presence and the amount of poorly differentiated areas within the tumour.
Our patient was more than 25 years old, had a tumour of more than 5 cm with abundant necrosis and poorly differentiated histology, a non-extremity site location and an SYT-SSX1 mutation. All these factors indicate an unfavourable prognostic outcome and can probably explain the aggressive behaviour of the tumour.
In summary, in the presence of unfavourable prognostic factors, neoadjuvant chemotherapy could be the primary approach to reduce the tumour size and the risk of distant micro-metastases and allowing a less aggressive radical surgery if the tumour is located in a non-extremity site. Hence, a multidisciplinary approach, even if having no influence on overall survival and disease-free survival, may improve the quality of life. In fact, in our patient we obtained complete clinical control in the pelvis with neoadjuvant chemotherapy, so avoiding pelvic exenteration.
References
1. Pelosi G, Luzzatto F, Landoni F, Staffa N, Maggioni A, Braidotti P, Cabras A, Aiello A, Del Curto B and Viale G (2007) Poorly differentiated synovial sarcoma of the vagina: first reported case with immunohistochemical, molecular and ultrastructural data Histopathology 50 809–10 doi 10.1111/j.1365-2559.2007.02647.x
2. Enzinger FM and Weiss SW (ed) (1995) Soft Tissue Tumours 3rd edn (St Louis, MO: Mosby) pp 757–86
3. Lamovec J, Zidar A and Cucek-Plenicar M (1988) Synovial sarcoma associated with total hip replacement: A case report J Bone Joint Surg 70 1558–60 PMID 3058710
4. Mitsuhashi A, Nagai Y, Suzuka K, Yamazawa K, Nojima T, Nikaido T, Ishikura H, Matsui H and Shozu M (2007) Primary synovial sarcoma in fallopian tube: case report and literature review Int J Gynecol Pathol 26 34–7 PMID 17197895 doi 10.1097/01.pgp.0000225841.13880.3a
5. Holloway CL, Russell AH, Muto M, Albert M and Viswanathan AN (2007) Synovial cell sarcoma of the vulva: multimodality treatment incorporating preoperative external-beam radiation, hemivulvectomy, flap reconstruction, interstitial brachytherapy, and chemotherapy Gynecol Oncol 104 253–6 PMID 17070900 doi 10.1016/j.ygyno.2006.09.018
6. Ambani DS, White B, Kaplan AL and Alberto A (2006) A case of monophasic synovial sarcoma presenting as a vulvar mass Gynecol Oncol 100 433–6 PMID 16226798 doi 10.1016/j.ygyno.2005.09.013
7. Nielsen GP, Shaw PA, Rosenberg AE, Dickersin GR, Young RH and Scully RE (1996) Synovial sarcoma of the vulva: a report of two cases Mod Pathol 9 970–4 PMID 8902833
8. Okagaki T, Ishida T, Hilgers RD (1976) A malignant tumour of the vagina resembling synovial sarcoma: a light and electron microscopic study Cancer 37 2306–20 PMID 56985 doi 10.1002/1097-0142(197605)37:53.0.CO;2-O
9. de Bruijn D, Nap J, and van Kessel A (2007) The (epi)genetics of human synovial sarcoma Genes Chromosomes Cancer 46 107–9 PMID 17117414
10. Kawai A, Woodruff J, Healey JH et al (1998) SYT-SSX gene fusion as a determinant of morphology and prognosis in synovial sarcoma N Engl J Med 338 153–60 PMID 9428816 doi 10.1056/NEJM199801153380303
11. Ladanyi M, Antonescu CR, Leung DH et al (2002) Impact of SYT-SSX fusion type on the clinical behavior of synovial sarcoma: a multi-institutional retrospective study of 243 patients Cancer Res 62 135–40 PMID 11782370
12. van der Heide HJ, Veth RP, Pruszczynski M, Wobbes T, van Hoesel QG and Lemmens JA (1998) Synovial sarcoma: Oncological and functional results Eur J Surg Oncol 24 114–9 PMID 9591026 doi 10.1016/S0748-7983(98)91433-0
13. Bergh P, Meis-Kindblom JM, Gherlinzoni F, Berlin O, Bacchini P, Bertoni F, Gunterberg B and Kindblom LG (1999) Synovial sarcoma: Identification of low and high risk groups Cancer 85 2596–607 PMID 10375108 doi 10.1002/(SICI)1097-0142(19990615)85:123.0.CO;2-K
14. Andrassy RJ, Okcu MF, Despa S and Raney RB (2001) Synovial sarcoma in children: Surgical lessons from a single institution and review of the literature J Am Coll Surg 192 305–13 PMID 11245372 doi 10.1016/S1072-7515(00)00806-1
15. Ladenstein R, Treuner J and Koscielniak E (1993) Synovial sarcoma of childhood and adolescence: report of the German CWS-81 study Cancer 71 3647–55 PMID 8387883 doi 10.1002/1097-0142(19930601)71:113.0.CO;2-U
16. Ferrari A, Casanova M and Massimino M (1999) Synovial sarcoma: report of a series of 25 consecutive children from a single institution Med Pediatr Oncol 32 32–7 PMID 9917750 doi 10.1002/(SICI)1096-911X(199901)32:13.0.CO;2-1
17. Raney R (2005) Synovial Sarcoma in young people: background, prognostic factors, and therapeutic questions J Pediatr Hematol Oncol 27 207–11 PMID 15838392 doi 10.1097/01.mph.0000161764.60798.60
18. Wright PH, Sim FH and Soule EH (1982) Synovial sarcoma J Bone Joint Surg Am 64 112–22 PMID 6274877
19. Mullen JR and Zagars GK (1994) Synovial sarcoma-outcome following conservation surgery and radiotherapy Radiother Oncol 33 23–30 PMID 7878206 doi 10.1016/0167-8140(94)90082-5
20. Oda Y, Hashimoto H and Tsuneyoshi M (1993) Survival in synovial sarcoma. A multivariate study of prognostic factors with special emphasis on the comparison between early death and long term survival. Am J Surg Pathol 17 35–44 PMID 8383468
21. Paulino AC (2004) Synovial sarcoma prognostic factors and patterns of failure Am J Clin Oncol 27 122–7 PMID 15057149 doi 10.1097/01.coc.0000047130.91699.DC
22. Hajdu SI, Shiu MH and Fortner JG (1977) Tendosynovial sarcoma: a clinicopathological study of 136 cases Cancer 39 1201–17 PMID 199345 doi 10.1002/1097-0142(197703)39:33.0.CO;2-P
23. Cadman NL, Soule EH and Kelly PJ (1965) Synovial sarcoma: An analysis of 134 tumours Cancer 15 613–27 doi 10.1002/1097-0142(196505)18:53.0.CO;2-V
24. Brodsky JT, Burt ME, Hajdu SI, Casper ES and Brennan MF (1992) Tendosynovial sarcoma. Clinicopathologic features, treatment, and prognosis. Cancer 70 484–9 PMID 1319818
25. Chandu de Silva M, McMahon A and Reid R (2004) Prognostic factors associated with local recurrence, metastases, and tumour-related death in patients with synovial sarcoma Am J Clin Oncol 27 113–21 PMID 15057148 doi 10.1097/01.coc.0000047129.97604.D6