Women’s cancers: how the discovery of BRCA genes is driving current concepts of cancer biology and therapeutics
Pooja Murthy1,2 and Franco Muggia1
1New York University School of Medicine, New York, NY 10016, USA
2Maimonides Cancer Center, Brooklyn, NY 11220, USA
Correspondence to: Pooja Murthy. Email: pmurthy@maimonidesmed.org
Abstract
Over the last two decades, discoveries related to the breast cancer susceptibility genes 1 and 2 (BRCA1 and BRCA2) have profoundly changed our understanding and management of hereditary breast and ovarian cancers. The concept of synthetic lethality, which arises when cells become vulnerable to a combination of deficiencies in DNA repair, has driven the expanding roles of poly (adenosine diphosphate (ADP)-ribose) polymerase inhibitors in breast and ovarian cancers, and prevention strategies are taking into account the tissue specificity, natural history (fallopian tube origin of some high-grade serous ovarian cancers) and hormone sensitivity of BRCA-associated cancers. Current research has focussed on further elucidating the roles of BRCA proteins in DNA repair, investigating other key DNA repair processes and proteins and linking aberrant DNA repair with carcinogenesis. The ultimate goal is to translate this evolving knowledge into improving the clinical care and treatment of patients with pathogenic BRCA variants or other deficiencies in homologous recombination (HR). In this review, we will discuss 1) the role of BRCA proteins in DNA repair; 2) emerging concepts in the biology of HR deficiency and 3) implications for prevention and treatment.
Keywords: ovarian cancer, breast cancer, BRCA, BRCA1, BRCA2, homologous recombination, DNA repair, DNA damage response, high-grade serous ovarian cancer, triple negative breast cancer, PARP inhibitor, synthetic lethality, bilateral salpingo-oophorectomy, prophylactic mastectomy, chemoprevention, immunotherapy
Copyright: © the authors; licensee ecancermedicalscience. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Published: 14/02/2019; Received: 17/09/2018
Introduction and historical landmarks
Family studies first by Henry Lynch and then by others pointed to a genetic origin for familial breast and ovarian cancers, with lifetime risks exceeding 50% in such family members. Interventions to diminish these risks by prophylactic surgeries or by drugs aiming to diminish hormonal actions on the breast (using surrogate endpoints such as breast density) were introduced once the hereditary nature of this predisposition was defined. Mary-Claire King’s genetic studies indicated a locus on chromosome 17p for a putative susceptibility gene. Shortly thereafter, identification and cloning of the breast cancer susceptibility genes 1 and 2 (BRCA1 and BRCA2) [1, 2] took place and led to a marked expansion of our knowledge on genetic susceptibility, its function and impact on tumour biology, and both preventive and therapeutic interventions. Soon after the cloning of the BRCA genes, Livingston’s laboratory and others found that loss of BRCA function was associated with a defect in homologous recombination (HR), a critical DNA repair pathway [3, 4]. Mouse models replicated the consequences of silencing BRCA genes in the development of triple negative breast cancer and demonstrated the effectiveness of DNA damaging agents such as platinum agents in their treatment. More recently, these studies have given way to the concept of synthetic lethality, which arises when cells become vulnerable to a combination of deficiencies in DNA repair, whereas a deficiency in only one pathway may not be lethal, thereby allowing for tumour-specific toxicity. This concept drove the initial development of poly (ADP-ribose) polymerase (PARP) inhibitors in the seminal phase I study of the PARP inhibitor olaparib in patients with advanced ovarian cancer and germline BRCA mutations [5]. The development of additional agents targeting PARP in other oncologic areas, both as single agents and in combination with other drugs, stalled for some time but eventually culminated in the 2014 Food and Drug Administration (FDA) approval of olaparib for the treatment of patients with deleterious or suspected deleterious germline BRCA-mutated advanced ovarian cancer who had progressed on three or more lines of chemotherapy [Lynparza prescribing information, 2014].
Table 1 features the highlights of conferences relevant to BRCA biology over the past 15 years.
Cancer risk overview and epidemiology
Hereditary breast and ovarian cancers are primarily related to highly penetrant germline mutations in either one of the two breast cancer susceptibility genes, BRCA1 and BRCA2 [6–8]. Carriers are heterozygous in one germline allele, and cancer may develop with loss of the wild type allele. Among women with ovarian cancer, regardless of family history, approximately 15% carry germline BRCA mutations [9]. In the general population of women with breast cancer in Western countries, 4%–5% carry deleterious BRCA mutations [10, 11], increasing to 12% in women who are less than 40 years old at the time of diagnosis [12]. Prevalence rates are also high among certain ethnicities; 10%–12% of breast cancers in the Ashkenazi Jewish population, unselected for family history, are attributable to mutations in BRCA1 or BRCA2 [13].
The cancer risks for patients with one of the three germline founder mutations in BRCA1 and BRCA2 have been extensively described. Similar information has gradually emerged for BRCA1 and BRCA2 variants across various ethnicities. One prospective cohort study evaluating over 9,000 mutation carriers, the majority from Europe, found the cumulative breast cancer risk to age 80 years was 72% for BRCA1 and 69% for BRCA2 carriers. The cumulative ovarian cancer risk to age 80 years was 44% for BRCA1 and 17% for BRCA2 carriers [14]. The overall risk of pancreatic cancer is about 1% and 4.9% for BRCA1 and BRCA2 mutation carriers, respectively [15, 16]. The prostate cancer risk is also increased and may range from 9% in BRCA1 mutation carriers to 33% in BRCA2 mutation carriers [17, 18]. Risks for melanoma, skin cancer, other gastrointestinal cancers and endometrial cancer may also be increased, but are not well characterised, and are often found with advancing age in individuals who have been successfully treated earlier for either breast or ovarian cancer [18]. Less is known about cancer risks for rarer mutations such as PALB2 and other genes that relate to the HR pathway.
Much remains to be learned about the prevalence of pathogenic BRCA variants in unselected patients with breast and ovarian cancers. The initially noted high penetrance of BRCA mutations has contributed to the ongoing interest in studying the specific pathogenesis, management and treatment issues for this subset of patients.
Table 1. Tracking our progress in BRCA genes: symposia on prevention and treatment.
Overview of DNA repair and roles of BRCA and related genes
Repair of DNA damage
DNA repair processes are essential in maintaining genetic integrity. The repair of a double-stranded DNA break (DSB) is particularly critical since an unresolved DSB often leads to genetic instability and cell death [19]. DSBs can be repaired through one of four possible mechanisms: nonhomologous end joining (NHEJ), HR, single-strand annealing (SSA) and microhomology-mediated end joining (MMEJ).
In NHEJ, the DSB is joined through blunt end ligation [20, 21]. Although this pathway has the potential to restore the original DNA sequence, any processing of the ends prior to ligation through nucleases or polymerases can lead to deletion or insertion mutations [22]. NHEJ is consequently error-prone but is evolutionarily conserved perhaps due to its fast kinetics, relative independence from the cell cycle and mechanistic flexibility [21].
Instead of NHEJ, the DSB can be initially resected, leaving two 3’ single-stranded overhanging ends. Then, the break can be repaired through one of three possible mechanisms: SSA, MMEJ and HR. SSA relies on the presence of a homologous sequence on either side of the break. After the creation of the 3’ overhanging ends, these homologous sequences anneal to each other and the intervening sequences are deleted. The break is thus repaired but there is a loss of genetic information [23]. MMEJ is an error-prone DSB repair mechanism that uses short homologous sequences (microhomology) flanking the DSB to align the ends for repair and is associated with deletions, insertions and chromosomal translocations [24]. Some of the distinctions between SSA and MMEJ include different mediators of synapsis and differences in the length of the annealing intermediate (i.e. MMEJ uses very short homologous sequences whereas SSA uses longer stretches of homology) [25].
HR is the DSB repair process in which one of the strands from the DSB invades into a homologous template, preferentially the identical sister chromatid [26], and uses the homologous sequence as a template for nascent DNA. Since HR allows for faithful replication and repair based on the homologous sequence, the process generally preserves the reading frame, and in most cases, is error-free. This is in contrast to the error-prone NHEJ, SSA and MMEJ pathways.
The question regarding how a cell ‘chooses’ between the different DSB repair pathways in a specific biological context is beyond the scope of this review. The decision is certainly multifactorial, but many of the details have yet to emerge. Briefly, the stage of the cell cycle, availability of the sister chromatid, the balance of mediators of synapsis (including BRCA 1 and 2, RAD51, PARP, DNA polymerase theta, REV7, 53BP1, ataxia telangiectasia mutated (ATM) and many other key proteins), the chromatin context adjacent to the break, the nature of the break itself and tissue-specific factors all likely play a significant role [25, 27–30].
BRCA proteins in homologous recombination
HR is distinguished by its error-free repair of DSBs and is the predominant and promoted DSB repair process in the S/G2 phases of the cell cycle. The steps of HR have been reviewed in other articles [31–33], but we will present a brief, somewhat simplified overview of HR in mammalian cells here, highlighting the roles of BRCA1 and BRCA2.
Initially, ATM and ATM and RAD3-related (ATR) recognise the DSB and phosphorylate downstream targets. BRCA1, along with BARD1 and BRIP1, act as a scaffold to organise other proteins, including the Mre11-Rad50-Nbs1 (MRN) complex and C-terminal binding protein interacting protein (CtIP), which then facilitate end resection [34–36]. Antagonising mediators, such as 53BP1 and RIF1, may inhibit end resection at DSBs [29, 37, 38], shuttling the DSB into the NHEJ repair process instead. The end resection is extensive, in contrast to the less extensive end resection that can lead to the MMEJ pathway. Phosphorylated replication protein A binds to the resected end, protecting it and also facilitating the next step—Rad51 loading [39].
Strand invasion, the defining step of HR, may then occur. BRCA1 promotes the recruitment of BRCA2 to the newly end resected-DSB through the bridging protein PALB2 [40, 41]. BRCA2 then helps load the RAD51 strand-exchange protein complex onto the overhanging 3’ break end. Next, the RAD51 single-stranded-DNA filament invades the homologous DNA strand, which, in most cases, is the sister chromatid. The remainder of the repair occurs with the use of the sister chromatid as the homologous template [31–33].
BRCA2 and BRCA1 also function in another critical process: the protection of stalled replication forks. More specifically, BRCA1 and components of the Fanconi anaemia pathway may cooperate with BRCA2 in preventing nascent DNA degradation at stalled replication forks [42]. Some studies have shown that this process of fork stabilisation may be independent of HR [43]. Broadly speaking, emerging data point to a complex interplay of HR factors in fork remodelling and stability [44].
Additionally, it is important to note that BRCA1 and BRCA2 are involved in processes other than HR and DNA repair; recent studies have found that the proteins play important roles in cell cycle progression, transcriptional control, mammary development and mitosis itself [45, 46].
Phenotypic expression of homologous recombination deficiency (HRD)
HRD biomarkers—Cells become deficient in HR through loss of BRCA function or loss of function of any other protein/factor involved in the process. Assays for HRD have focussed on the detection of underlying driver mutations (ATM, alpha thalassemia/mental retardation syndrome X-linked (ATRX), BARD1, Bloom’s syndrome gene product (BLM), BRIP1, FANCA/C/D2/E/F/G/L, MRE11A, nibrin (NBN), PALB2, phosphatase and tensin homolog (PTEN), RAD50, RAD51, RAD51B and Werner syndrome helicase (WRN)), epigenetic changes such as BRCA promoter methylation, or the resultant mutational landscape of deficient HR (copy number alterations, structural rearrangements, including telomeric allelic imbalance, large-scale transition and loss of heterozygosity (LOH)) [47]. Head-to-head comparisons of the different HRD assays have not yet been done, and so it is not clear which assay is superior. However, the two most clinically validated HRD assays are the Myriad Genetics myChoice HRD Plus assay and the Foundation Medicine’s FoundationFocus companion diagnostic (CDx) xBRCA LOH assay. The Myriad myChoice assay measures telomeric allelic imbalance, large-scale transition and LOH. FoundationFocus CDxBRCA LOH is an assay that uses next-generation sequencing for the qualitative detection of BRCA1 and BRCA2 sequence alterations and LOH.
A few prospective clinical trials in breast and ovarian cancer have used these assays not only to stratify patients with or without HRD but also as inclusion or exclusion criteria. Although showing that HRD assays may have predictive value for response to platinum chemotherapy and PARP inhibitors, their ultimate role in selecting patients that may benefit from these drugs is unclear [48–51], especially as the indications for PARP inhibitors have broadened beyond patients with germline BRCA mutations to include patients with HRD from other causes and even to other ovarian cancers without identifiable defects in DNA repair. Additional data on the clinical use of HRD assays may emerge in the future.
Most of these assays focus on genomic signatures or scars, which may or may not represent the tumor’s current HR capability. Cells with HRD may in time regain their ability to perform HR through reversion mutations or other mechanisms, and therefore, most of the current assays are limited in identifying such tumours since they may be unable to capture these new mutations. Functional assays for HRD should be able to overcome these limitations but remain in the research arena since they require fresh tissue.
HRD and tissue-specific tumour development—An unanswered question in BRCA-related cancer biology is why HR defects are associated with cancer development both in terms of tissue specificity (breast and ovary) and histology (i.e. high-grade serous predominance).
BRCA-mutation-related sex hormone alterations have been sought to explain carcinogenesis in hormone-sensitive tissues such as the breast and ovary. BRCA1 can interact with oestrogen and progesterone receptors to decrease sex hormone transcription [52], and premenopausal women with germline BRCA mutations have been found to have higher serum progesterone levels in the luteal phase of the menstrual cycle than non-carriers [53]. Mice models of BRCA1-deficient breast tumours have shown increased progesterone receptor expression in both the breast tumours and the adjacent benign tissue [54], and that mammary tumorigenesis can be prevented with progesterone inhibition [55]. The receptor activator of nuclear factor kappa-B ligand (RANKL) is an important paracrine mediator of progesterone signalling in breast tissues. Preclinical studies have shown that RANK-RANKL signalling is augmented in a population of luminal progenitor cells from precancerous breast tissues with heterozygous BRCA1 mutations, and that pharmacological inhibition of the RANK/RANKL pathway delayed the onset and reduced incidence of breast tumours in mice models [56–58]. Therefore, inhibition of the RANK-RANKL pathway has become an active focus of the clinical investigation on the prevention of BRCA-associated breast cancers.
Oestrogen may also play a role; oestrogen can overcome HRD-related oxidative stress-induced cell death through induction of the transcription factor NRF2, which regulates an antioxidant pathway [59, 60]. Oestrogen may act through the PI3K - protein kinase B (PKB, also known as AKT) - mammalian target of rapamycin (mTOR) pathway to induce NRF2 accumulation [59, 61], supporting the emerging clinical strategy of treating BRCA1-related cancers with PI3K inhibitors. Targeting the PI3K-AKT-mTOR pathway has established a basis in breast cancer treatment. Everolimus, an mTOR inhibitor, is approved and has demonstrated activity in combination with exemestane in hormone receptor-positive, HER2-negative breast cancer [62]. Most recently, the PI3K inhibitor alpelisib also showed a progression-free survival benefit in hormone receptor-positive, HER2-negative breast cancer [63].
Fallopian tube origin of BRCA-mutation driven high-grade serous ovarian cancers may also reflect the contribution of oestrogen-dependent fimbrial proliferation in their pathogenesis, beginning with TP53 immunostaining and the serous tubal intraepithelial cancer (STIC) [64, 65].
Haploinsufficiency—Patients with germline BRCA mutations are heterozygous at the mutated BRCA allele and their heterozygous cells are assumed to be biologically normal until alteration of this unmutated allele occurs, resulting in loss of BRCA functionality. More recently, Pathania et al. [66] showed that human mammary epithelial cells and fibroblasts with heterozygous BRCA mutations (BRCA1mut/ ) are intrinsically ‘haploinsufficient’ in BRCA-dependent stalled fork repair when compared to mammary epithelial cells and fibroblasts without BRCA mutations (BRCA1 / ). When subjected to ultraviolet (UV) radiation, heterozygous cells exhibited multiple defects of stalled fork repair resulting in increased replication stress and DNA breaks compared to wild type cells, while retaining proficiency in other BRCA1 functions such as mitosis, cell cycle control, mammary development and heterochromatin-based satellite RNA suppression. HR-mediated repair of double-stranded breaks, while unaltered by infrared radiation, was demonstrably impaired if replication stress was induced via increasing exposure to UV followed by infrared irradiation—as assessed by RAD51 recruitment and olaparib cytotoxicity in comparison to wild type BRCA cells. This ‘conditional’ haploinsufficiency in HR may be expected to occur when seemingly normal cells are subjected to a certain threshold of replication stress [66]. Interestingly, Winqvist et al. [67] found that B lymphocytes and T cells from PALB2 heterozygotes were also haploinsufficient for replication stress responsiveness, suggesting that the defect could extend to mutations in other genes associated with BRCA function. At present, one can only speculate that such deficient stalled fork repair may lead to tumorigenesis: could secondary squamous cell carcinomas of the oral cavity in BRCA-mutated patients exposed to radiation or certain types of chemotherapy, or skin cancers and melanomas in excessively sun-exposed subjects be manifestations of haploinsufficiency?
Prevention and treatment of BRCA-mutated ovarian and related gynaecologic cancers: current status
Prevention
Individuals with a germline pathogenic BRCA1 or BRCA2 mutation should be counselled at the time of disclosure of the test results and options for primary prevention should be discussed. Unfortunately, there is a lack of high-quality data showing survival benefits for ovarian cancer screening in women with pathogenic BRCA mutations. However, screening with concurrent transvaginal ultrasound and CA-125 is commonly used in mutation carriers who have not undergone bilateral salpingo-oophorectomy (BSO) and is suggested as an option in the current National Comprehensive Cancer Network (NCCN) guidelines [68].
Over the past two decades, risk-reducing surgeries for ovarian cancer arising in BRCA1 and BRCA2 mutation carriers have gradually strengthened the concept that most high-grade serous ovarian cancers arise in the fimbria of the fallopian tubes and go through a premalignant stage (identified by p53 immunostaining), an intraepithelial neoplasia stage (serous tubal intraepithelial carcinomas or STICs), and then the development of invasive carcinoma [69]. Occasionally the tubes and ovaries are totally fused within a neoplastic mass at the time of surgery, and often the cancer is already disseminated throughout the peritoneal cavity at diagnosis.
Given the limitations of screening tests and the poor outcomes associated with advanced high-grade serous ovarian cancers, the focus has been on preventive strategies for women at high risk for breast and ovarian cancer, including BRCA mutation carriers. Risk-reducing BSO is recommended for women with pathogenic BRCA mutations and who have completed childbearing, either by age 35–40 or individualised based on onset of ovarian cancer in family members, based on studies that have found that BSO significantly reduces both the risk of ovarian cancer (by 70%–80%) and all-cause mortality in BRCA mutation carriers [70, 71] and may also have a protective effect on breast cancer risk if completed prior to menopause [70, 72]. It is important to note that a continued, small risk of primary peritoneal cancer persists after BSO.
A major clinical issue is when to institute risk-reducing surgeries in premenopausal women who are not ready for BSO, either because they are contemplating childbearing or for other reasons. Since a majority of high-grade serous ovarian cancers may arise from the fallopian tubes, salpingectomy with delayed oophorectomy is an alternative potential strategy for these patients and is currently being studied in the women choosing surgical prevention clinical trial (NCT02760849).
Other preventive measures include the use of oral contraceptives for ovarian cancer risk reduction. A meta-analysis of six studies evaluating ovarian cancer risk in BRCA 1/2 mutation carriers and eight studies evaluating breast cancer risk in BRCA 1/2 mutation carriers found an inverse association between oral contraceptive use and ovarian cancer risk (odds ratio, 0.58; 95% confidence interval (CI), 0.46–0.73), but a non-statistically significant trend towards an increased breast cancer risk (odds ratio, 1.21; 95% CI, 0.93–1.58) [73]. Other data assessing oral contraceptive use and breast cancer risk in BRCA mutation carriers have been conflicting [74, 75]. Thus, women with BRCA mutations should be carefully counselled on the benefits and potential harms of oral contraceptives prior to using them as an alternative way to reduce the risk of ovarian cancer.
Even though deleterious mutations in both BRCA1 and BRCA2 result in defects in HR and predispose patients to develop breast and ovarian cancers, most breast cancers that develop in BRCA1 mutation carriers are triple negative, whereas most breast cancers in BRCA2 mutation carriers are hormone receptor-positive, HER2-negative. This difference affects patient counselling since preventative measures may have more or less of an impact depending on whether the patient is a carrier of BRCA1 or BRCA2. For example, BRCA2 carriers may have more of a reduction in breast cancer risk with risk-reducing salpingo-oophorectomy than BRCA1 carriers based on a prospective multicentre cohort study [70] although another study found no significant reduction in risk of first breast cancer with BSO, even in BRCA2 carriers.
Treatment
The treatment of BRCA-mutated advanced ovarian cancer is distinguished from the general treatment of advanced ovarian cancer (without germline BRCA mutations), mainly through the inclusion of PARP inhibitors into management plans. A complete discussion of PARP inhibitors in ovarian cancer is beyond the scope of this review. We briefly summarise what is provided by Physician Data Query (PDQ) in www.cancer.gov.
Two PARPis are currently FDA-approved for the single-agent treatment of women with BRCA mutations and recurrent ovarian cancer: olaparib and rucaparib. Olaparib is approved for use in women with recurrent ovarian cancer and a germline BRCA mutation after three or more prior lines of treatment, and rucaparib is indicated for use in women with recurrent ovarian cancer and a known BRCA mutation (either somatic or germline) after two or more prior lines of treatment.
Phase 2 trials of olaparib and rucaparib demonstrated excellent objective response rates and progression-free survival, with better outcomes in patients with platinum-sensitive disease compared to patients with platinum-resistant disease [5, 76–78]. Olaparib also demonstrates activity in platinum-resistant ovarian cancer, as seen in a phase 2 basket trial of patients with germline BRCA 1/2 mutations and recurrent cancer. In the cohort of 193 patients with platinum-resistant ovarian cancer, 31% achieved an objective response [79].
The other three PARPs, niraparib, veliparib and talazoparib, are also being evaluated in advanced ovarian cancer in monotherapy and combination therapy trials. Ongoing combination therapy trials are assessing PARPis with chemotherapy, immunotherapy and targeted agents, including PI3-kinase inhibitors, ATM inhibitors and angiogenesis inhibitors.
Three PARPis are currently FDA-approved for the maintenance treatment of patients with platinum-sensitive relapsed ovarian cancer who had a partial or complete response to platinum-based chemotherapy, regardless of BRCA status: niraparib, olaparib and rucaparib. Approval was based on randomised trials that demonstrated a progression-free survival benefit to PARP inhibition compared to placebo [80–82]. Subgroup analyses from these trials demonstrated a greater benefit for patients with germline BRCA mutations compared to patients without germline BRCA mutations [80]. Most recently, olaparib was approved in the first line maintenance setting for patients with advanced ovarian cancer and pathogenic BRCA mutations, following the results of a phase 3 randomised trial that showed that 2-year consolidation resulted in a significantly improved progression-free survival that was sustained after drug discontinuation, at a median follow-up of 36 months [83].
Prevention and treatment of BRCA-mutated breast cancer: current status
Prevention
Risk-reducing prophylactic bilateral mastectomy has been shown to decrease the incidence of breast cancer by more than 90% in women at risk of hereditary breast cancer [70]. The NCCN, therefore, recommends that prophylactic bilateral mastectomy be offered to BRCA carriers [68]. However, in contrast to ovarian cancer, effective screening tests (mammogram and MRI) for breast cancer exist and so the choice to undergo prophylactic bilateral mastectomy should be individualised to the patient and based on her personal preferences. As mentioned previously, risk-reducing BSO may reduce the risk of breast cancer in BRCA mutation carriers if completed prior to menopause, although the extent of breast cancer risk reduction has varied considerably between studies [70, 72].
Current non-surgical prevention strategies are mainly limited to chemoprevention with tamoxifen, a selective oestrogen receptor modulator. There is insufficient data addressing the preventive benefit of aromatase inhibitors or raloxifene for patients with BRCA mutations although several trials have shown that these drugs reduce the risk of breast cancer in the general population of higher risk women. The data supporting tamoxifen use in BRCA mutation carriers are also relatively limited; subset analyses of a National Surgical Adjuvant Breast and Bowel Project Breast Cancer Prevention trial (P-1 trial) demonstrated a reduction in breast cancer risk with tamoxifen use for patients with pathogenic BRCA mutations [84]. The preventive benefit of tamoxifen was mainly noted in BRCA2 mutation carriers and not in patients with BRCA1 mutations, likely because BRCA2-mutated breast cancer tends to be hormone receptor-positive. Even though tamoxifen may primarily reduce the risk of hormone receptor-positive breast cancer, prospective studies of mutation carriers with a diagnosis of breast cancer and who took tamoxifen showed a reduction in contralateral breast cancers, including a significant reduction in BRCA1-associated breast cancers, suggesting that tamoxifen may also reduce the risk for triple negative breast cancers [85, 86]. Overall, data supporting tamoxifen use in BRCA mutation carriers are more limited than chemoprevention data for the general population of high-risk women. Tamoxifen can be considered for women who decline prophylactic mastectomies, especially patients with BRCA2 mutations, but patients should understand that risk-reducing prophylactic mastectomies are more effective in reducing breast cancer risk.
Other strategies for chemoprevention of both BRCA-associated breast and ovarian cancers are being actively evaluated in preclinical and clinical studies. As discussed previously, RANKL inhibition holds particular promise and is being evaluated in a pilot clinical trial (NCT03382574). Letrozole, recombinant human chorionic gonadotropin and lifestyle changes are also being evaluated as preventive measures for BRCA-associated breast cancer in clinical trials (NCT00673335, NCT03495609 and NCT02516540).
Treatment
The treatment of patients with advanced breast cancer and deleterious BRCA mutations was not appreciably different from the treatment of patients with wild-type BRCA status and advanced breast cancer until studies indicated that patients with BRCA mutated-breast cancer were especially sensitive to treatment with platinum chemotherapy. The largest of these studies was the phase 3 Triple Negative Trial (TNT) trial, which randomised patients with advanced triple negative breast cancer to treatment with either carboplatin or docetaxel. In the unselected population, there was no significant difference in activity between the two therapies, but in patients with germline BRCA mutations, carboplatin demonstrated a significantly longer progression-free survival and overall response rate compared to docetaxel (overall response rate 68% versus 33%, respectively, p = 0.01) [87].
PARP inhibitors were then evaluated in advanced BRCA-mutated breast cancer, following compelling results from ovarian cancer clinical trials. OlympiAD was a randomised phase 3 trial, in which olaparib monotherapy was compared with standard physician’s choice chemotherapy (capecitabine, vinorelbine or eribulin) in patients with germline BRCA mutations and metastatic HER2-negative breast cancer. The progression-free survival was 7.0 months in the olaparib group and 4.2 months in the physician’s choice chemotherapy group (hazard ratio 0.58, p < 0.001) [88]. The larger phase 3 EMBRACA trial evaluated talazoparib (a PARPi with potent PARP trapping ability) versus physician’s choice chemotherapy in a similar patient population with HER2-negative advanced breast cancer and deleterious germline BRCA 1/2 mutations, and similarly found a longer progression-free survival in the talazoparib arm with acceptable toxicity (8.6 months versus 5.6 months, p < 0.0001) [89]. There was no significant difference in overall survival in the OlympiAD trial (the trial was not powered to detect a difference), and overall survival results are not yet mature for the EMBRACA trial. In January 2018, following the results from OlympiAD, the FDA approved olaparib for the treatment of patients with deleterious or suspected deleterious germline BRCA-mutated, HER2-negative metastatic breast cancer and who had been previously treated with chemotherapy either in the neoadjuvant, adjuvant or metastatic setting. Talazoparib subsequently gained FDA approval in October 2018 for patients with deleterious or suspected deleterious germline BRCA-mutated, HER2-negative locally advanced or metastatic breast cancer.
The unresolved question from these trials is how to fit PARP inhibitors into the treatment scheme for patients with germline BRCA mutations and advanced breast cancer, and specifically, how to sequence platinum chemotherapy with PARP inhibitors. Subgroup analyses from OlympiAD and EMBRACA show that in patients who had previously received platinum chemotherapy, response rates to the PARP inhibitors were lower than other subgroups. The phase 2 ABRAZO trial evaluated talazoparib in patients with advanced breast cancer and germline BRCA mutations and demonstrated a progression-free survival of 4 months for patients who had progressed at least 8 weeks after the last dose of platinum chemotherapy. In sum, although the response to PARP inhibition may be lower in patients previously treated with platinum chemotherapy compared to platinum-naïve patients, PARP inhibitors still demonstrated efficacy in patients who had responded to platinum chemotherapy in the metastatic setting or had relapsed at least a few months after treatment.
An alternate strategy would be to treat early in the metastatic disease course with PARP inhibition and then follow with platinum chemotherapy at progression. A phase 2 trial of patients with germline BRCA 1/2-mutated metastatic breast cancer assessed single-agent veliparib, another PARPi, followed by veliparib plus carboplatin at disease progression. The post-progression treatment with veliparib and carboplatin at the maximum tolerated doses (150 mg twice per day and area under the curve (AUC) of 5, respectively) yielded minimal benefit; only one patient out of 30 had a response [90]. Overall, the optimum sequence of therapy in patients with germline BRCA-mutated metastatic breast cancer still needs to be determined.
An ongoing randomised, prospective, phase 2 clinical trial is comparing cisplatin with or without veliparib in patients with metastatic triple negative and/or BRCA-mutated breast cancer (NCT02595905). A hypothesis is that combination therapy, if tolerated, could be more effective than sequential therapy since it may attenuate the development of cross-resistance. Overlap between the mechanisms of platinum and PARP inhibitor resistance is being explored. However, a recurring issue in trials combining PARP inhibitors with chemotherapy is the dose-limiting toxicity, particularly myelosuppression. Most of the combination trials with chemotherapy and PARP inhibitors have dealt with the toxicities by prioritising the chemotherapeutic agent and using lower doses of the PARP inhibitor, such as in the phase 3 BrighTNess trial, which showed no improvement in pathologic complete response rate with the addition of veliparib to neoadjuvant carboplatin and paclitaxel in triple negative breast cancer [91]. Managing these toxicities and finding effective doses of both the PARP inhibitor and the chemotherapy agent remain significant challenges for chemotherapy and PARP inhibitor combination trials.
In terms of the treatment of BRCA-mutated early stage breast cancer, neoadjuvant or adjuvant platinum-based chemotherapy is commonly used, as supported by a few studies showing that platinum-based neoadjuvant chemotherapy leads to improved rates of pathological complete response [92, 93]. Of course, platinum-based chemotherapy is not appropriate for all patients (elderly, poor performance status and low-risk early-stage cancers) given the additional toxicity. These studies are also not definitive since they did not show improvements in disease-free and overall survival, were not restricted to patients with BRCA mutations or were not large randomised studies. However, when taken with the compelling results from the TNT trial, platinum-based neoadjuvant or adjuvant chemotherapy is favoured for BRCA-mutated patients. PARP inhibitors have not yet shown consistent activity in the neoadjuvant setting [91] and are not presently indicated in the neoadjuvant or adjuvant treatment of BRCA mutated-early stage breast cancer. However, a few PARPis are currently in phase 2–3 clinical trials assessing adjuvant or neoadjuvant therapy in women with HER2-negative breast cancer and germline BRCA 1/2 mutations.
Emerging therapeutic concepts in targeting HRD
This growing understanding of how BRCA mutations drive carcinogenesis has led to new concepts in the prevention, diagnosis and treatment of breast, ovarian and related gynaecologic cancers.
PARP inhibitors and PARP inhibitor (PARPi) resistance
The PARP family of enzymes functions by transferring ADP-ribose from nicotinamide adenine dinucleotide (NAD) to other proteins and is involved in a number of DNA repair processes. PARPis have demonstrated clinical activity in BRCA mutated breast and ovarian cancer and are thought to induce synthetic lethality in these tumours through the interaction between the HRD from BRCA deficiency and four PARPi-associated effects: defective base excision repair, PARP trapping on damaged DNA (PARP1 is unable to disassociate from DNA leading to obstructed replication forks), defective BRCA1 recruitment and activation of NHEJ [94].
Despite the demonstrated efficacy of PARP inhibition in BRCA-mutated breast and ovarian cancers, most patients eventually develop resistance, as evidenced by a median progression-free survival of less than 1 year in the major PARPi clinical trials. Several mechanisms of PARPi resistance have been described and include 1) increased PARPi drug efflux; 2) reversion mutations in BRCA or other mutated genes that restore protein function and thus restore HR; 3) increased NAD synthesis which mitigates PARP trapping; 4) loss of 53BP1 and REV7 and 5) replication fork stabilisation [95]. The development of reversion mutations that restore HR may be the most well-known mechanism of PARPi resistance [96]. Loss of p53-binding protein 1 (53BP1) is another significant PARPi resistance mechanism. 53BP1 acts together with RIF1 and inhibits the end resection step of HR, antagonising the function of BRCA1. Correspondingly, loss of 53BP1 has been shown to restore HR, even in cells with BRCA deficiency [29, 97].
Strategies to circumvent PARP inhibitor resistance are a major focus of preclinical research and clinical trials. One general strategy is an attempt at augmenting synthetic lethality by targeting other DNA damage repair mechanisms in combination with PARP inhibition, such as the ATR or ATM pathways. A few ongoing clinical trials are using this approach in both advanced breast and ovarian cancers (clinicaltrials.gov).
Recent studies have demonstrated that tumours deficient in HR up-regulate the MMEJ DNA repair pathway as a survival mechanism [30]. In vitro studies using HR-deficient ovarian cancer cell lines found that knockdown of MMEJ reduced the survival of the cells after exposure to both PARP and ATM inhibition [98]. Thus, the increase in the activity of the MMEJ pathway could be a way for tumours to maintain viability while receiving PARP inhibition, leading to PARPi resistance. This concept needs to be validated through further preclinical studies.
Immunotherapy
Recently, deficiencies in DNA repair processes have emerged as predictive biomarkers for response to immune checkpoint blockade. In ovarian cancer, tumour specimens from patients with high-grade serous ovarian carcinoma and BRCA1 or BRCA2 loss (through germline or somatic mutations, or BRCA methylation) had significantly increased immune cell infiltrates [99] and overexpression of PD-L1 [100] compared to high-grade serous ovarian cancer specimens without BRCA mutations. Recently, the preliminary results from TOPACIO/Keynote-162 were presented at the American Society of Clinical Oncology (ASCO) annual meeting in June 2018. This is a phase 2 trial evaluating the combination of niraparib, a PARPi, and pembrolizumab in platinum-resistant ovarian cancer. The primary endpoint was overall response rate, which was 25% in all patients and 45% in patients with BRCA mutations [101]. Follow-up is ongoing.
In breast cancer, a preclinical study using breast cancer cell lines showed that PARP inhibition up-regulated PD-L1 expression, and that combination treatment with PARP inhibition and anti-PD-L1 therapy was significantly more efficacious than either treatment alone [102]. The TOPACIO/Keynote-162 study in triple negative breast cancer was also presented at the recent ASCO 2018 annual meeting. This phase 2 study evaluated niraparib plus pembrolizumab in patients with metastatic triple negative breast cancer, of which 22% had deleterious BRCA mutations and 41% had received platinum chemotherapy in the metastatic setting. The overall response rate for all patients was 29% and was significantly higher in the patients with BRCA mutations [103]. Follow-up for this study is ongoing.
Teo et al. [104] evaluated 60 patients with advanced urothelial cancer from prospective clinical trials featuring nivolumab or atezolizumab and found that 25% of these patients had likely deleterious DNA damage repair mutations. These patients had a markedly better overall response rate compared to patients without DNA damage repair mutations (80% versus 19%, p < 0.001).
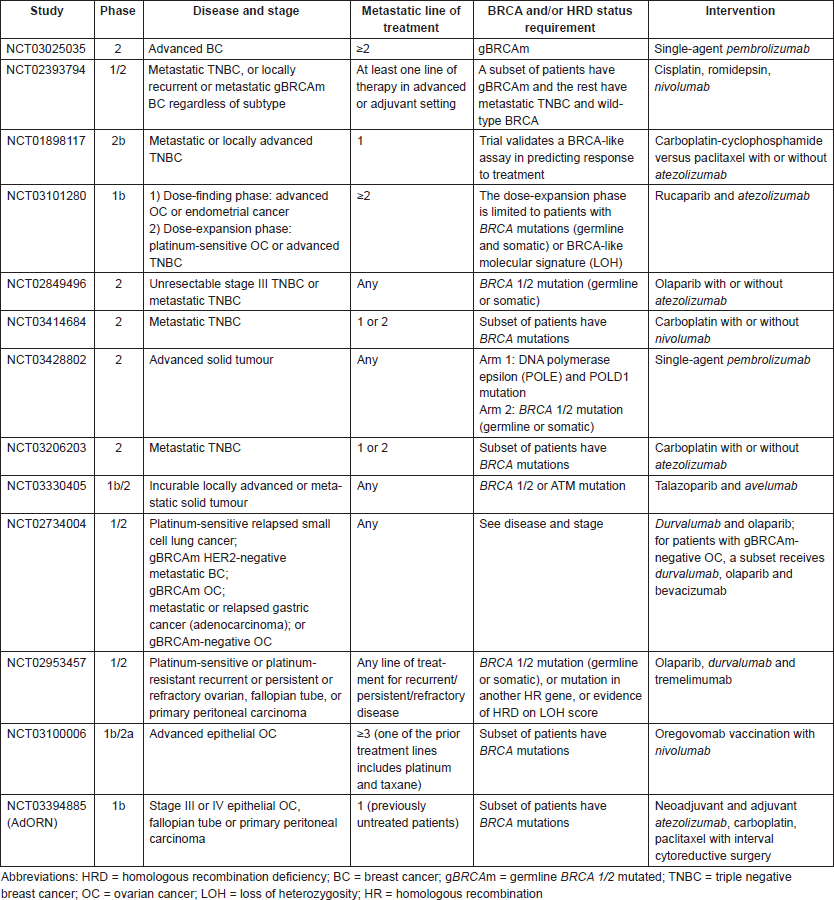
Taken together, immune checkpoint inhibition appears to have promising activity in tumours with HRD, including breast and ovarian cancers. Combination therapies with immune checkpoint inhibitors and agents targeting DNA damage repair are particularly intriguing since the neoantigen load may be further increased with drugs targeting DNA repair (such as PARPis) leading to a more immunogenic tumour. Multiple ongoing or planned clinical trials are evaluating immune checkpoint inhibitors alone or in combination with other drugs in breast and ovarian cancers with HRD (see Table 2 below).
Table 2. Ongoing clinical trials with immunotherapy in BRCA-like breast and ovarian cancer.
Conclusions
The cloning and discovery of the BRCA 1/2 genes over two decades ago drove a body of research on the biology of BRCAness that continues to expand today. Advances in understanding the interplay of DNA repair processes and the interaction between the DNA damage response and other host factors, such as the immune system and hormone regulation, are being translated into clinical trials in ovarian and breast cancer. Our growing knowledge of HRD is being developed into biomarkers for treatment response, and several assays for HRD are undergoing clinical validation. The role of PARP inhibition is also expanding. PARPis are being evaluated in new and earlier settings, such as first-line maintenance therapy and platinum-resistant disease in ovarian cancer and in neoadjuvant therapy in breast cancer. Combination therapies of PARPis with other agents are being evaluated in breast and ovarian cancer clinical trials, as a way to circumvent PARP inhibitor resistance, augment synthetic lethality and increase response. Increasingly, we recognise the heterogeneity of patients with BRCA mutations and breast or ovarian cancer and appreciate that many different driver processes may be at play in tumorigenesis. Thus, the future of BRCA-like breast and ovarian cancer treatment, like other cancers, may feature a designer therapeutic approach, in which we query which pathways are active in a specific tumour and target those.
Funding
The authors did not receive any funding for the preparation of this article.
Conflicts of interest
The authors declare no conflict of interest.
References
1. Miki Y, Swensen J, and Shattuck-Eidens D, et al (1994) A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1 Science 266(5182) 66–71 https://doi.org/10.1126/science.7545954 PMID: 7545954
2. Wooster R, Bignell G, and Lancaster J, et al (1995) Identification of the breast cancer susceptibility gene BRCA2 Nature 378(6559) 789 https://doi.org/10.1038/378789a0 PMID: 8524414
3. Snouwaert JN, Gowen LC, and Latour AM, et al (1999) BRCA1 deficient embryonic stem cells display a decreased homologous recombination frequency and an increased frequency of non-homologous recombination that is corrected by expression of a brca1 transgene Oncogene 18(55) 7900 https://doi.org/10.1038/sj.onc.1203334
4. Moynahan ME, Pierce AJ, and Jasin M (2001) BRCA2 is required for homology-directed repair of chromosomal breaks Mol cell 7(2) 263–272 https://doi.org/10.1016/S1097-2765(01)00174-5 PMID: 11239455
5. Fong PC, Yap TA, and Boss DS, et al (2010) Poly (ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval J clin oncol 28(15) 2512–2519 https://doi.org/10.1200/JCO.2009.26.9589 PMID: 20406929
6. Antoniou A, Cunningham A, and Peto J, et al (2008) The BOADICEA model of genetic susceptibility to breast and ovarian cancers: updates and extensions Br J cancer 98(8) 1457 https://doi.org/10.1038/sj.bjc.6604305 PMID: 18349832 PMCID: 2361716
7. Ford D, Easton D, and Stratton M, et al (1998) Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families Amer J Human Gen 62(3) 676–689 https://doi.org/10.1086/301749
8. Peto J, Collins N, and Barfoot R, et al (1999) Prevalence of BRCA1 and BRCA2 gene mutations in patients with early-onset breast cancer J Nat Can Inst 91(11) 943–949 https://doi.org/10.1093/jnci/91.11.943
9. Alsop K, Fereday S, and Meldrum C (2012) BRCA mutation frequency and patterns of treatment response in BRCA mutation–positive women with ovarian cancer: a report from the Australian Ovarian Cancer Study Group J Clin Oncol 30(21) 2654 https://doi.org/10.1200/JCO.2011.39.8545 PMID: 22711857 PMCID: 3413277
10. Papelard H, De Bock G, and Van Eijk R, et al (2000) Prevalence of BRCA1 in a hospital-based population of Dutch breast cancer patients Br J Cancer 83(6) 719 https://doi.org/10.1054/bjoc.2000.1331 PMID: 10952774 PMCID: 2363536
11. Malone KE, Daling JR, and Doody DR, et al (2006) Prevalence and predictors of BRCA1 and BRCA2 mutations in a population-based study of breast cancer in white and black American women ages 35 to 64 years Cancer Res 66(16) 8297–8308 https://doi.org/10.1158/0008-5472.CAN-06-0503 PMID: 16912212
12. Copson ER, Maishman TC, and Tapper WJ, et al (2018) Germline BRCA mutation and outcome in young-onset breast cancer (POSH): a prospective cohort study Lancet Oncol https://doi.org/10.1016/S1470-2045(17)30891-4 PMCID: 5805863
13. Warner E, Foulkes W, and Goodwin P, et al (1999) Prevalence and penetrance of BRCA1 and BRCA2 gene mutations in unselected Ashkenazi Jewish women with breast cancer J Nat Cancer Inst 91(14) 1241–1247 https://doi.org/10.1093/jnci/91.14.1241 PMID: 10413426
14. Kuchenbaecker KB, Hopper JL, and Barnes DR, et al (2017) Risks of breast, ovarian, and contralateral breast cancer for BRCA1 and BRCA2 mutation carriers Jama 317(23) 2402–2416 https://doi.org/10.1001/jama.2017.7112 PMID: 28632866
15. Thompson D and Easton DF (2002) Cancer incidence in BRCA1 mutation carriers J Nat Can Inst 94(18) 1358–1365 https://doi.org/10.1093/jnci/94.18.1358
16. Ferrone CR, Levine DA, and Tang LH, et al (2009) BRCA germline mutations in Jewish patients with pancreatic adenocarcinoma J Clin Oncol 27(3) 433 https://doi.org/10.1200/JCO.2008.18.5546
17. Leongamornlert D, Mahmud N, and Tymrakiewicz M, et al (2012) Germline BRCA1 mutations increase prostate cancer risk BrJ Cancer 106(10) 1697 https://doi.org/10.1038/bjc.2012.146
18. Consortium BCL (1999) Cancer risks in BRCA2 mutation carriers J Nat Can Inst 91(15) 1310–1316 https://doi.org/10.1093/jnci/91.15.1310
19. Huertas P (2010) DNA resection in eukaryotes: deciding how to fix the break Nat Struc Mol Biol 17(1) 11 https://doi.org/10.1038/nsmb.1710
20. Chiruvella KK, Liang Z, and Wilson TE (2013) Repair of double-strand breaks by end joining Cold Spring Harbor Perspec Biol 5(5) a012757 https://doi.org/10.1101/cshperspect.a012757
21. Lieber MR (2010) The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway Ann Rev Biochem 79 181–211 https://doi.org/10.1146/annurev.biochem.052308.093131 PMID: 20192759 PMCID: 3079308
22. Bétermier M, Bertrand P, and Lopez BS (2014) Is non-homologous end-joining really an inherently error-prone process? PLoS Gen 10(1) e1004086 https://doi.org/10.1371/journal.pgen.1004086
23. Heyer W-D, Ehmsen KT, and Liu J (2010) Regulation of homologous recombination in eukaryotes Ann Rev Gen 44 113–139 https://doi.org/10.1146/annurev-genet-051710-150955
24. Sfeir A and Symington LS (2015) Microhomology-mediated end joining: a back-up survival mechanism or dedicated pathway? Trends Biochem Sci 40(11) 701–714 https://doi.org/10.1016/j.tibs.2015.08.006 PMID: 26439531 PMCID: 4638128
25. Bhargava R, Onyango DO, and Stark JM (2016) Regulation of single-strand annealing and its role in genome maintenance Trends Gen 32(9) 566–575 https://doi.org/10.1016/j.tig.2016.06.007
26. Johnson RD and Jasin M (2000) Sister chromatid gene conversion is a prominent double-strand break repair pathway in mammalian cells EMBO J 19(13) 3398–3407 https://doi.org/10.1093/emboj/19.13.3398 PMID: 10880452 PMCID: 313931
27. Stark JM, Pierce AJ, and Oh J, et al (2004) Genetic steps of mammalian homologous repair with distinct mutagenic consequences Mol Cell Biol 24(21) 9305–9316 https://doi.org/10.1128/MCB.24.21.9305-9316.2004 PMID: 15485900 PMCID: 522275
28. Tutt A, Bertwistle D, and Valentine J, et al (2001) Mutation in Brca2 stimulates error-prone homology-directed repair of DNA double-strand breaks occurring between repeated sequences EMBO J 20(17) 4704–4716
29. Isono M, Niimi A, and Oike T, et al (2017) BRCA1 directs the repair pathway to homologous recombination by promoting 53BP1 dephosphorylation Cell Rep 18(2) 520–532 https://doi.org/10.1016/j.celrep.2016.12.042 PMID: 28076794
30. Ceccaldi R, Rondinelli B, and D’Andrea AD (2016) Repair pathway choices and consequences at the double-strand break Trends Cell Biol 26(1) 52–64 https://doi.org/10.1016/j.tcb.2015.07.009 PMCID: 4862604
31. Prakash R, Zhang Y, and Feng W, et al (2015) Homologous recombination and human health: the roles of BRCA1, BRCA2, and associated proteins Cold Spring Harbor Persp Biol 7(4) a016600 https://doi.org/10.1101/cshperspect.a016600
32. Jasin M and Rothstein R (2013) Repair of strand breaks by homologous recombination Cold Spring Harbor Persp Biol 5(11) https://doi.org/10.1101/cshperspect.a012740
33. Li X and Heyer W-D (2008) Homologous recombination in DNA repair and DNA damage tolerance Cell Res 18(1) 99 https://doi.org/10.1038/cr.2008.1 PMID: 18166982 PMCID: 3087377
34. Mimitou EP and Symington LS (2008) Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing Nature 455(7214) 770 https://doi.org/10.1038/nature07312 PMID: 18806779 PMCID: 3818707
35. Zhu Z, Chung W-H, and Shim EY, et al (2008) Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends Cell 134(6) 981–994 https://doi.org/10.1016/j.cell.2008.08.037 PMID: 18805091 PMCID: 2662516
36. Sartori AA, Lukas C, and Coates J, et al (2007) Human CtIP promotes DNA end resection Nature 450(7169) 509 https://doi.org/10.1038/nature06337 PMID: 17965729 PMCID: 2409435
37. Cao L, Xu X, and Bunting SF, et al (2009) A selective requirement for 53BP1 in the biological response to genomic instability induced by Brca1 deficiency Mol cell 35(4) 534–541 https://doi.org/10.1016/j.molcel.2009.06.037 PMID: 19716796 PMCID: 3392030
38. Di Virgilio M, Callen E, and Yamane A, et al (2013) Rif1 prevents resection of DNA breaks and promotes immunoglobulin class switching Science 1230624
39. Park MS, Ludwig DL, and Stigger E, et al (1996) Physical interaction between human RAD52 and RPA is required for homologous recombination in mammalian cells J Biol Chem 271(31) 18996–19000 https://doi.org/10.1074/jbc.271.31.18996 PMID: 8702565
40. Xia B, Sheng Q, and Nakanishi K, et al (2006) Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2 Molecular Cell 22(6) 719–729 https://doi.org/10.1016/j.molcel.2006.05.022 PMID: 16793542
41. Sy SM, Huen MS, and Chen J (2009) PALB2 is an integral component of the BRCA complex required for homologous recombination repair Pro Nat Acad Sci 106(17) 7155–7160 https://doi.org/10.1073/pnas.0811159106
42. Schlacher K, Wu H, and Jasin M (2012) A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2 Cancer Cell 22(1):106-16 https://doi.org/10.1016/j.ccr.2012.05.015 PMID: 22789542 PMCID: 3954744
43. Schlacher K, Christ N, and Siaud N, et al (2011) Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11 Cell 145(4) 529–542 https://doi.org/10.1016/j.cell.2011.03.041 PMID: 21565612 PMCID: 3261725
44. Mijic S, Zellweger R, and Chappidi N, et al (2017) Replication fork reversal triggers fork degradation in BRCA2-defective cells Nat Commun8(1) 859 https://doi.org/10.1038/s41467-017-01164-5 PMID: 29038466 PMCID: 5643541
45. Lee H (2014) Cycling with BRCA2 from DNA repair to mitosis Exp Cell Res 329(1) 78–84
46. Choi E, Park P-G, and Lee H-O, et al (2012) BRCA2 fine-tunes the spindle assembly checkpoint through reinforcement of BubR1 acetylation Dev Cell 22(2) 295–308 https://doi.org/10.1016/j.devcel.2012.01.009 PMID: 22340495
47. Brok WDD, Schrader KA, and Sun S, et al (2017) Homologous recombination deficiency in breast cancer: a clinical review JCO Pre Oncol 1 1–13
48. Von Minckwitz G, Timms K, and Untch M, et al (2015) Prediction of pathological complete response (pCR) by homologous recombination deficiency (HRD) after carboplatin-containing neoadjuvant chemotherapy in patients with TNBC: results from GeparSixto Am Soc Clin Oncol
49. Schouten PC, Marme F, and Aulmann S, et al (2014) Breast cancers with a BRCA1-like DNA copy number profile recur less often than expected after high-dose alkylating chemotherapy Clin Cancer Res Clincan 1894
50. Isakoff SJ, Mayer EL, and He L, et al (2015) TBCRC009: a multicenter phase II clinical trial of platinum monotherapy with biomarker assessment in metastatic triple-negative breast cancer J clin Oncol 33(17) 1902–1999 https://doi.org/10.1200/JCO.2014.57.6660 PMID: 25847936 PMCID: 4451173
51. Graeser MK, McCarthy A, and Lord CJ, et al (2010) A marker of homologous recombination predicts pathological complete response to neoadjuvant chemotherapy in primary breast cancer Clin Can Res Clincan 1027
52. Katiyar P, Ma Y, and Fan S, et al (2006) Regulation of progesterone receptor signaling by BRCA1 in mammary cancer Nucl Recept Signal 4 https://doi.org/10.1621/nrs.04006 PMID: 16741564 PMCID: 1472667
53. Widschwendter M, Rosenthal AN, and Philpott S, et al (2013) The sex hormone system in carriers of BRCA1/2 mutations: a case-control study Lancet Oncol 14(12) 1226–1232
54. King TA, Gemignani ML, and Li W, et al (2004) Increased progesterone receptor expression in benign epithelium of BRCA1-related breast cancers Cancer Res 64(15) 5051–5053 https://doi.org/10.1158/0008-5472.CAN-04-1283 PMID: 15289302
55. Poole AJ, Li Y, and Kim Y, et al (2006) Prevention of Brca1-mediated mammary tumorigenesis in mice by a progesterone antagonist Science 314(5804) 1467–1470 https://doi.org/10.1126/science.1130471 PMID: 17138902
56. Sigl V, Owusu-Boaitey K, and Joshi PA, et al (2016) RANKL/RANK control Brca1 mutation-driven mammary tumors Cell Res 26(7) 761 https://doi.org/10.1038/cr.2016.69 PMID: 27241552 PMCID: 5129883
57. Nolan E, Lindeman GJ, and Visvader JE (2017) Out-RANKing BRCA1 in mutation carriers Cancer Res https://doi.org/10.1158/0008-5472.CAN-16-2025 PMID: 28104682
58. Nolan E, Vaillant F, and Branstetter D, et al (2016) RANK ligand as a potential target for breast cancer prevention in BRCA1-mutation carriers Nat Med 22(8) 933 https://doi.org/10.1038/nm.4118 PMID: 27322743
59. Gorrini C, Gang BP, and Bassi C, et al (2014) Estrogen controls the survival of BRCA1-deficient cells via a PI3K–NRF2-regulated pathway Proc Nat Acad Sci 201324136
60. Gorrini C, Baniasadi PS, and Harris IS, et al (2013) BRCA1 interacts with Nrf2 to regulate antioxidant signaling and cell survival J Exp Med 210(8) 1529–1544 https://doi.org/10.1084/jem.20121337 PMID: 23857982 PMCID: 3727320
61. Mitsuishi Y, Taguchi K, and Kawatani Y, et al (2012) Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming Cancer Cell 22(1) 66–79 https://doi.org/10.1016/j.ccr.2012.05.016 PMID: 22789539
62. Baselga J, Campone M, and Piccart M, et al (2012) Everolimus in postmenopausal hormone-receptor–positive advanced breast cancer N Engl J Med 366(6) 520–529 https://doi.org/10.1056/NEJMoa1109653
63. André F, Ciruelos E, and Rubovszky G, et al (2018) LBA3_PR Alpelisib (ALP) fulvestrant (FUL) for advanced breast cancer (ABC): Results of the phase III SOLAR-1 trial Ann Oncol 29(8) https://doi.org/10.1093/annonc/mdy424.010
64. Dubeau L and Drapkin R (2013) Coming into focus: the nonovarian origins of ovarian cancer Ann Oncol 24(8) viii28–viii35 https://doi.org/10.1093/annonc/mdt308 PMID: 24131966 PMCID: 3805308
65. Kuhn E, Kurman RJ, and Vang R, et al (2012) TP53 mutations in serous tubal intraepithelial carcinoma and concurrent pelvic high-grade serous carcinoma—evidence supporting the clonal relationship of the two lesions J Pathol 226(3) 421–426 https://doi.org/10.1002/path.3023
66. Pathania S, Bade S, and Le Guillou M, et al (2014) BRCA1 haploinsufficiency for replication stress suppression in primary cells Nat Commun 5 5496 https://doi.org/10.1038/ncomms6496 PMID: 25400221 PMCID: 4243249
67. Nikkilä J, Parplys AC, and Pylkäs K, et al (2013) Heterozygous mutations in PALB2 cause DNA replication and damage response defects Nat Commun 4 2578 https://doi.org/10.1038/ncomms3578 PMID: 24153426 PMCID: 3826652
68. Network NCC (2019) Genetic/familial high-risk assessment: breast and ovarian (version 2, 2019) [Available from: https://www.nccn.org/professionals/physician_gls/pdf/genetics_screening.pdf]
69. Labidi-Galy SI, Papp E, and Hallberg D, et al (2017) High grade serous ovarian carcinomas originate in the fallopian tube Nat Commun 8(1) 1093 https://doi.org/10.1038/s41467-017-00962-1 PMID: 29061967 PMCID: 5653668
70. Domchek SM, Friebel TM, and Singer CF, et al (2010) Association of risk-reducing surgery in BRCA1 or BRCA2 mutation carriers with cancer risk and mortality Jama 304(9) 967–975 https://doi.org/10.1001/jama.2010.1237 PMID: 20810374 PMCID: 2948529
71. Finch AP, Lubinski J, and Møller P, et al (2014) Impact of oophorectomy on cancer incidence and mortality in women with a BRCA1 or BRCA2 mutation J Clin Oncol 32(15) 1547 https://doi.org/10.1200/JCO.2013.53.2820 PMID: 24567435 PMCID: 4026578
72. Heemskerk-Gerritsen B, Seynaeve C, and Van Asperen C, et al (2015) Breast cancer risk after salpingo-oophorectomy in healthy BRCA1/2 mutation carriers: revisiting the evidence for risk reduction JNCI 107(5) https://doi.org/10.1093/jnci/djv033
73. Moorman PG, Havrilesky LJ, and Gierisch JM, et al (2013) Oral contraceptives and risk of ovarian cancer and breast cancer among high-risk women: a systematic review and meta-analysis J Clin Oncol 31(33) 4188–4198 https://doi.org/10.1200/JCO.2013.48.9021 PMID: 24145348
74. Lee E, Ma H, and McKean-Cowdin R, et al (2008) Effect of reproductive factors and oral contraceptives on breast cancer risk in BRCA1/2 mutation carriers and noncarriers: results from a population-based study Cancer Epidemiol Prev Biomarkers 17(11) 3170–3178 https://doi.org/10.1158/1055-9965.EPI-08-0396
75. Narod SA, Dubé M-P, and Klijn J, et al (2002) Oral contraceptives and the risk of breast cancer in BRCA1 and BRCA2 mutation carriers J Nat Cancer Inst 94(23) 1773–1779 https://doi.org/10.1093/jnci/94.23.1773 PMID: 12464649
76. Domchek SM, Aghajanian C, and Shapira-Frommer R, et al (2016) Efficacy and safety of olaparib monotherapy in germline BRCA1/2 mutation carriers with advanced ovarian cancer and three or more lines of prior therapy Gynecol Oncol 140(2) 199–203 https://doi.org/10.1016/j.ygyno.2015.12.020 PMID: 26723501 PMCID: 4992984
77. Audeh MW, Carmichael J, and Penson RT, et al (2010) Oral poly (ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial Lancet 376(9737) 245–251 https://doi.org/10.1016/S0140-6736(10)60893-8 PMID: 20609468
78. Kristeleit RS, Shapira-Frommer R, and Oaknin A, et al (2016) Clinical activity of the poly (ADP-ribose) polymerase (PARP) inhibitor rucaparib in patients (pts) with high-grade ovarian carcinoma (HGOC) and a BRCA mutation (BRCAmut): Analysis of pooled data from Study 10 (parts 1, 2a, and 3) and ARIEL2 (parts 1 and 2) Ann Oncol 27(6) https://doi.org/10.1093/annonc/mdw374.03
79. Kaufman B, Shapira-Frommer R, and Schmutzler RK, et al (2014) Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation J Clin Oncol 33(3) 244–250 https://doi.org/10.1200/JCO.2014.56.2728 PMID: 25366685 PMCID: 6057749
80. Mirza MR, Monk BJ, and Herrstedt J, et al (2016) Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer N Engl J Med 375(22) 2154–2164 https://doi.org/10.1056/NEJMoa1611310 PMID: 27717299
81. Ledermann J, Harter P, and Gourley C, et al (2012) Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer New England J Med 366(15) 1382–1392 https://doi.org/10.1056/NEJMoa1105535
82. Coleman RL, Oza AM, and Lorusso D, et al (2017) Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial Lancet 390(10106) 1949–1961 https://doi.org/10.1016/S0140-6736(17)32440-6 PMID: 28916367 PMCID: 5901715
83. Moore K, Colombo N, and Scambia G, et al (2018) Maintenance Olaparib in patients with newly diagnosed advanced ovarian cancer New England J Med https://doi.org/10.1056/NEJMoa1810858
84. King M-C, Wieand S, and Hale K, et al (2001) Tamoxifen and breast cancer incidence among women with inherited mutations in BRCA1 and BRCA2: national surgical adjuvant breast and bowel project (NSABP-P1) breast cancer prevention trial Jama 286(18) 2251–2256 https://doi.org/10.1001/jama.286.18.2251 PMID: 11710890
85. Gronwald J, Tung N, and Foulkes WD, et al (2006) Tamoxifen and contralateral breast cancer in BRCA1 and BRCA2 carriers: an update Int J Cancer 118(9) 2281–2284 https://doi.org/10.1002/ijc.21536
86. Phillips K-A, Milne RL, and Rookus MA, et al (2013) Tamoxifen and risk of contralateral breast cancer for BRCA1 and BRCA2 mutation carriers J Clin Oncol 31(25) 3091 https://doi.org/10.1200/JCO.2012.47.8313 PMID: 23918944 PMCID: 3753701
87. Tutt A, Tovey H, and Cheang MCU, et al (2018) Carboplatin in BRCA1/2-mutated and triple-negative breast cancer BRCAness subgroups: the TNT Trial
88. Robson M, Im S-A, and Senkus E, et al (2017) Olaparib for metastatic breast cancer in patients with a germline BRCA mutation N Engl J Med 377(6) 523–533 https://doi.org/10.1056/NEJMoa1706450 PMID: 28578601
89. Litton JK, Rugo HS, and Ettl J, et al (2017) A phase 3 trial comparing talazoparib, an oral PARP inhibitor, to physician’s choice of therapy in patients with advanced breast cancer and a germline BRCA-mutation San antonio Breast Cancer Symposium
90. Somlo G, Frankel PH, and Arun BK, et al (2017) Efficacy of the PARP inhibitor veliparib with carboplatin or as a single agent in patients with germline BRCA1-or BRCA2-associated metastatic breast cancer: california cancer consortium trial NCT01149083 Clin Can Res 23(15) 4066–4076 https://doi.org/10.1158/1078-0432.CCR-16-2714
91. Loibl S, O’Shaughnessy J, and Untch M, et al (2018) Addition of the PARP inhibitor veliparib plus carboplatin or carboplatin alone to standard neoadjuvant chemotherapy in triple-negative breast cancer (BrighTNess): a randomised, phase 3 trial Lan Oncol 19(4) 497–509 https://doi.org/10.1016/S1470-2045(18)30111-6
92. Byrski T, Gronwald J, and Huzarski T, et al (2009) Pathologic complete response rates in young women with BRCA1-positive breast cancers after neoadjuvant chemotherapy J Clin Oncol 28(3) 375–379 https://doi.org/10.1200/JCO.2008.20.7019 PMID: 20008645
93. Byrski T, Huzarski T, and Dent R, et al (2014) Pathologic complete response to neoadjuvant cisplatin in BRCA1-positive breast cancer patients Breast Cancer Res Treat 147(2) 401–405 https://doi.org/10.1007/s10549-014-3100-x PMID: 25129345
94. Konecny G and Kristeleit R (2016) PARP inhibitors for BRCA1/2-mutated and sporadic ovarian cancer: current practice and future directions Br J Cancer 115(10) 1157 https://doi.org/10.1038/bjc.2016.311 PMID: 27736844 PMCID: 5104889
95. Thomas A, Murai J, and Pommier Y (2018) The evolving landscape of predictive biomarkers of response to PARP inhibitors J Clin Invest 128(5) https://doi.org/10.1172/JCI120388
96. Barber LJ, Sandhu S, and Chen L, et al (2013) Secondary mutations in BRCA2 associated with clinical resistance to a PARP inhibitor J Pathol 229(3) 422–429 https://doi.org/10.1002/path.4140
97. Bunting SF, Callén E, and Wong N, et al (2010) 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks Cell 141(2) 243–254 https://doi.org/10.1016/j.cell.2010.03.012 PMID: 20362325 PMCID: 2857570
98. Ceccaldi R, Liu JC, and Amunugama R, et al (2015) Homologous-recombination-deficient tumours are dependent on Polθ-mediated repair Nature 518(7538) 258 https://doi.org/10.1038/nature14184 PMID: 25642963 PMCID: 4415602
99. McAlpine JN, Porter H, and Köbel M, et al (2012) BRCA1 and BRCA2 mutations correlate with TP53 abnormalities and presence of immune cell infiltrates in ovarian high-grade serous carcinoma Mod Pathol 25(5) 740 https://doi.org/10.1038/mod-pathol.2011.211 PMID: 22282309
100. Strickland KC, Howitt BE, and Shukla SA, et al (2016) Association and prognostic significance of BRCA1/2-mutation status with neoantigen load, number of tumor-infiltrating lymphocytes and expression of PD-1/PD-L1 in high grade serous ovarian cancer Oncotarget 7(12) 13587 https://doi.org/10.18632/oncotarget.7277 PMID: 26871470 PMCID: 4924663
101. Konstantinopoulos PA, Waggoner SE, and Vidal GA, et al (2018) TOPACIO/Keynote-162 (NCT02657889): A phase 1/2 study of niraparib pembrolizumab in patients (pts) with advanced triple-negative breast cancer or recurrent ovarian cancer (ROC)—Results from ROC cohort Am Soc Clin Oncol https://doi.org/10.1200/JCO.2018.36.15_suppl.106
102. Jiao S, Xia W, and Yamaguchi H, et al (2017) PARP inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppression Clin Can Res https://doi.org/10.1158/1078-0432.CCR-16-3215
103. Vinayak S, Tolaney SM, and Schwartzberg LS, et al (2018) TOPACIO/Keynote-162: Niraparib pembrolizumab in patients (pts) with metastatic triple-negative breast cancer (TNBC), a phase 2 trial Am Soc Clin Oncol https://doi.org/10.1200/JCO.2018.36.15_suppl.1011
104. Teo MY, Seier K, and Ostrovnaya I, et al (2018) Alterations in DNA damage response and repair genes as potential marker of clinical benefit from PD-1/PD-L1 blockade in advanced urothelial cancers J Clin Oncol 75 7740