Successful treatment of a stage IIIC small-cell carcinoma of the ovary hypercalcemic subtype using multi-modality therapeutic approach
Qian Qin1, Veronica B Ajewole2,4, Tiffany G Sheu3, Rachel Donohue3 and Monisha Singh2
1Department of Internal Medicine, Houston Methodist Hospital, Houston, TX 77030, USA
2Houston Methodist Cancer Centre, Houston Methodist Hospital, Houston, TX 77030, USA
3Department of Pathology and Genomic Medicine, Houston Methodist Hospital, Houston, TX 77030, USA
4Department of Pharmacy Practice, Texas Southern University College of Pharmacy and Health Sciences, Houston, TX 77004, USA
Correspondence to: Qian Qin. Email: qqin@houstonmethodist.org
Abstract
Small-cell carcinoma of the ovary, hypercalcemic type (SCCOHT) is a rare but highly undifferentiated, aggressive malignancy that primarily affects young women. Due to its early onset, unclear familial history and vague presenting symptoms, most SCCOHT patients present late with advanced disease. The prognosis is extremely poor, with <10% disease-free survival for advanced stages. Although several therapeutic regimens have been proposed, to date there is no consensus on the optimal strategy.
Here, we describe a successful case of advanced-stage SCCOHT of the left ovary treated with cytoreductive surgery, semi-intense chemotherapy, high-dose consolidative chemotherapy, autologous hematopoietic stem cell transplantation and pelvic radiation with long-term survival. Given the almost universal mortality of advanced SCCOHT in long-term follow-up, we believe this case highlights the importance of prompt diagnosis when a young patient presents with abdominal swelling and hypercalcemia as well as early, aggressive, combined modality treatment. This case is also especially remarkable given the patient underwent fertility preservation surgery, which is not recommended by most of the current literature. However, as therapies improve and more young patients may survive SCCOHT, the question of fertility will increase in relevance. We believe the pros and cons of conservation should be discussed in detail with the patient.
Keywords: small cell carcinoma of the ovary hypercalcemic subtype, SCCOHT, advance stage, therapy
Copyright: © the authors; licensee ecancermedicalscience. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Published: 03/05/2018; Received: 17/01/2018
Introduction
First described by Dickersin et al [1] in 1982, small-cell carcinoma of the ovary, hypercalcemic type (SCCOHT) is a rare but highly undifferentiated, aggressive malignancy [1]. Although the age at diagnosis ranges from 14 months to 71 years, it primarily affects young women with a median of 23 years [1–4]. Approximately ~60% of SCCOHT cases present with paraneoplastic hypercalcemia [2]. Due to its early onset, unclear familial history and vague presenting symptoms, most SCCOHT patients present late with advanced disease [2–4]. The prognosis is generally very poor, with a 33% disease-free survival for stage IA disease and exponentially poorer prognosis (<10%) in more advanced stages [2, 3]. Diagnosis is difficult owing to the unclear cell lineage that SCCOHT arises from and its histological similarity to a wide variety of tumours [2–6]. The disease’s tendency for rapid progression and high recurrence also makes treatment a challenge. Several therapeutic regimens have been proposed; however, there is no consensus on the optimal strategy [3, 7–10].
We describe a previously healthy 19-year-old female who was diagnosed with stage IIIC SCCOHT. She successfully underwent semi-intensive chemotherapy treatment, conditioning chemotherapy, autologous hematopoietic stem cell transplantation (HSCT), as well as pelvic/para-aortic radiation based on the novel regimen designed by Pautier et al [7] the only prospective trial to date treating 27 patients with SCCOHT [7]. Our patient remains disease free 8 years from initial diagnosis.
Case presentation
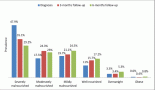
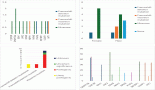
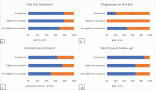
A previously healthy 19-year-old G0P0 female initially presented with several months history of increasing abdominal distension, weight loss, constipation and generalised malaise. Computerised tomography (CT) scan revealed a 20 cm mass extending from the left ovary. The patient was also tachycardic to the 120 s with a fever of 101.2 oF and mild hypotension. The patient had a negative pregnancy test, normal alpha fetoprotein level at 2.3 ng/mL, normal carcinoembryonic antigen at <0.5 ng/mL but an elevated cancer antigen (CA) 125 at 375 U/mL (normal <30 U/mL) raising concerns for gynaecological malignancy. The patient also had hypercalcemia with uncorrected serum calcium of 16 mg/dL, normal thyroid stimulating hormone, a low parathyroid hormone (PTH), but an elevated PTH related peptide level at 5.0 pmol/L. She underwent left salpingo-oophorectomy, omentectomy, bilateral pelvic lymph node dissection, bilateral para-aortic lymph node dissection and peritoneal biopsy. The right ovary and uterus were spared in an attempt to preserve endocrine and fertility functions. The resected tumour size was 20.5 cm, weight was 1602 g, with large areas of necrosis but negative resection margins and uninvolved left fallopian tube (Figure 1). One of the fourteen dissected lymph nodes was positive. Pathology slide revealed SCCOHT and the patient was diagnosed with stage IIIC disease (Figures 2 and 3). The patient reported no other personal or family histories of malignancies.
Given the aggressive nature of the disease and a lack of therapeutic consensus, the patient was initiated on a regimen based on that reported by Pautier et al [7] after correspondences with the author and tumour board discussion. She received six cycles of semi-intensive chemotherapy with cisplatin 100 mg/m2 and adriamycin 40 mg/m2 on day one, as well as etoposide 75 mg/m2 and cyclophosphamide 300 mg/m2 on days 1–3. The regimen was given for every 21 days with pegfilgrastim support. The patient experienced fatigue, nausea, vomiting and diarrhoea, but no symptoms above grade two. These were well controlled with antiemetic and antidiarrhoeal medications. A CT scan of the chest, abdomen and pelvis post chemotherapy showed no residual disease. Serum calcium, PTH related peptide and CA125 also normalised to 9.0 mg/dL, <0.2 pmol/L and 6 U/mL, respectively.
The patient was considered to have a complete response. A bone marrow biopsy showed no pathological involvement and the process for autologous HSCT was initiated. A total of 7.845 × 106 CD34/kg stem cells were collected after stem cell mobilisation with filgrastim. The patient subsequently underwent high-dose conditioning chemotherapy with carboplatin 400 mg/m2, etoposide 450 mg/m2 and cyclophosphamide 1600 mg/m2 followed by autologous HSCT. Her transplantation course was complicated by neutropenic fever requiring broad spectrum antibiotics, as well as odynophagia and severe mucositis with poor appetite requiring short-term total parenteral nutrition. Her symptoms slowly improved with full engraftment approximately 2 weeks post transplantation. She was discharged home in a stable condition. The patient subsequently completed 30 days of radiation therapy to the pelvic and para-aortic areas via intensity modulated radiation therapy with a cumulative dose of 40 Gy without complications. Her follow-up over the subsequent years showed no disease recurrence.
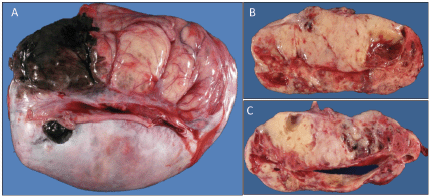
Figure 1. (a): Gross image of the external surface of the left ovary (20.5 cm, 1.6 kg). (b and c): representative cut surfaces of the left ovary.
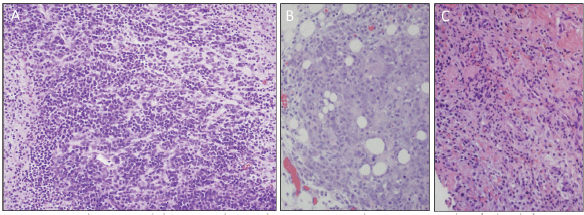
Figure 2. High-powered hematoxylin and eosin stain photomicrograph of the left ovary, omentum, and posterior cul de sac peritoneum (a, b and c, respectively) all showing diffuse sheet-like architecture of small round cells with scant cytoplasm, hyperchromatic nuclei and small nucleoli.
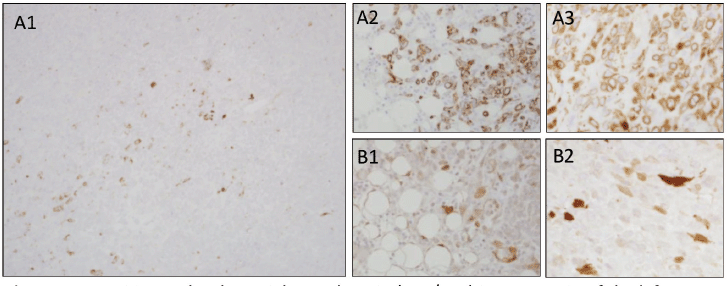
Figure 3. (a): Positive molecular weight cytokeratin (AE1/AE3) immunostain of the left ovary, omentum and posterior cul de sac peritoneum (a1, a2 and a3, respectively). (b): Positive calretinin immunostaining of the omentum and posterior cul de sac peritoneum (b1 and b2, respectively).
Discussion/literature review
Here, we describe a 19-year-old patient with stage IIIC SCCOHT who has remained disease free for 8 years since initial diagnosis. This is a remarkable outcome considering the unfavourable characteristics at diagnosis (tumour size >10 cm, age <30 years and elevated pre-operative calcium) and the advance disease stage. Most literature agrees upon the almost universal mortality of advanced SCCOHT in long-term follow-up [2, 3, 8, 11]. This extremely poor prognosis warrants closer evaluation of the clinical symptoms, pathological findings, treatment modalities and post-therapy surveillance of SCCOHT.
In the landmark study by Young et al [2], the clinical and pathological features of 150 patients with SCCOHT were analysed. The tumour is almost always unilateral with right-side predominance and a mean tumour size of 15 cm2. Patients usually presented with vague symptoms including subacute nausea and vomiting, abdominal pain, swelling, fatigue and lethargy similar to other ovarian malignancies. However, the median age of diagnosis for SCCO is 23 years with hypercalcemia observed in 62% of the patients [2]. These clinical findings have been documented in subsequent publications [3,11]. As such, a young patient presenting with a negative pregnancy test but abdominal swelling and hypercalcemia should raise concerns for SCCOHT.
The pathological basis of SCCOHT remains unclear but suggests a primitive germ cell origin. The initial histopathology described in 1982 reported neoplastic cells with structural similarities to those of epithelial nature. However, they lacked any of the specific identifying characteristics associated with germline, sex cord, Sertoli or granulosa cells [1]. The cells were simply described as diffuse infiltrates of small cells with scant cytoplasm and hyperchromatic nuclei, and the malignancy was deemed small-cell carcinoma of the ovary with histopathology to be determined [1]. The subsequent study by Ulbright et al [13] supports a germ cell origin, especially related to yolk sac tumours, based on similar epidemiological distributions as well as light microscopy, immunohistochemical, and ultrastructural examinations. A recent comprehensive literature review, whole tissue staining and tissue microarray of SCCOHT cases by McCluggage et al [14] further evidenced its primitive germ cell origin as many demonstrated SALL4 positivity. Recent studies also suggest germline or somatic deleterious mutation of SMARCA4 as a major propagator of SCCOHT [15–18]. Especially validating is the comprehensive genetic profiling by Lin et al [19] which revealed SMARCA4 inactivation in 94% of the analysed SCCOHT cases with 62.5% predicted to be germline mutations. The study also revealed very few re-occurring genetic alterations, further supporting SMARCA4 as the main driver in SCCOHT tumourgenesis [19].
Despite these advances, there is still a lack of consensus in the treatment of SCCOHT. In a retrospective study by Callegaro-Filho et al [11], the treatment and disease course of 47 patients with SCCOHT were analysed with varying surgical debulking, chemotherapy regimens, plus or minus radiation and autologous HSCT. Recurrent disease was unfortunately observed in 74.5% of the cases [11]. Given the correlation of SCCOHT to germ cell tumours and prominent data suggesting benefit of high-dose chemotherapy with autologous HSCT in germ cell tumours, the recent years have seen a move towards more intensive, multi-modality treatment regimens with higher successes [8, 9, 15]. Especially in Pautier et al [7], the only prospective study to date, the patients underwent nonconservative debulking surgery, semi-intensive chemotherapy (cisplatin, adriamycin, etoposide and cyclophosphamide), high-dose consolidation chemotherapy (carboplatin, etoposide and cyclophosphamide) followed by autologous HSCT [7]. The outcomes were better for early-stage patients although late-stage diseases remain difficult to manage [7]. In their concluding remarks, Pautier et al [7] recommended considering increasing doses of initial chemotherapy as well as the addition of pelvic radiation therapy to improve survival. We thus added radiation therapy to our regimen and believe this helped decrease local recurrence and improve our patient’s clinical outcome. Other currently explored modalities include targeted therapy with EZH2 inhibitors and immunotherapy with programmed death 1/programmed death-ligand 1 (PD-1/PD-L1) inhibitors [20, 21]. Specifically, the above-mentioned SMARCA4 alteration leads to BRG1 loss-of-function, which may allow for sensitivity to tazemetostat, an EZH2 inhibitor currently implemented in a phase II trial that includes SCCOHT (clinicaltrials.gov, NCT02601950) [19, 20]. PD-1 and PD-L1 inhibitors in turn may offset adaptive immune evasion of the SCCOHT tumour cells [21].
Finally, although SCCOHT is a disease with >95% unilateral ovarian involvement, most current literature argues against fertility conserving surgery [1, 7]. However, given the young population and long-lasting endocrine effects, bilateral oophorectomy should not be performed lightly. Especially as the above-mentioned therapies improve and more patients survive SCCOHT, the question of preservation will increase in relevance. Although very few cases exist on its success, our patient with stage IIIC disease underwent cytoreductive but fertility preserving surgery. The patient declined ovarian protection with gonadotropin-releasing hormone (GnRH) agonist or cryopreservation prior to chemotherapy; however, she now has the return of normal menstruation and is currently seeking fertility counselling to bear children [22, 23]. While a more definitive answer is difficult to achieve without a noninferiority trial, we believe the conversation of pros and cons regarding fertility conservation should be discussed with patients presenting with SCCOHT.
Conclusion
We have presented a successful case of advanced-stage SCCOHT of the left ovary treated with cytoreductive surgery but with uterus and contralateral ovary preservation, intense chemotherapy, high-dose consolidative chemotherapy, autologous HSCT and pelvic radiation with long-term survival. Due to its early onset, aggressive disease course and poor prognosis, we believe this early, high intensity, multimodality treatment regimen should be considered when approaching SCCOHT.
Conflicts of interest and funding statement
All authors have no conflict of interests and nothing to disclose. No funding was accepted for this case study.
References
1. Dickersin GR, Kline IW, and Scully RE (1982) Small cell carcinoma of the ovary with hypercalcemia: a report of 11 cases Cancer 49 188–197 PMID: 6274502
2. Young RH, Oliva E, and Scully RE (1994) Small cell carcinoma of the ovary, hypercalcemic type. A clinicopathological analysis of 150 cases Am J Surg Pathol 18 1102–1116
3. Estel R, Hackethal A, and Kalder M, et al (2011) Small cell carcinoma of the ovary of the hypercalcaemic type: an analysis of clinical and prognostic aspects of a rare disease on the basis of cases published in the literature Arch Gynecol Obstet 284 1277–1282 https://doi.org/10.1007/s00404-011-1846-5 PMID: 21298438
4. Ulbright TM, Roth LM, and Stehman FB, et al (1987) Poorly differentiated (small cell) carcinoma of the ovary in young women: evidence supporting a germ cell origin Hum Pathol 18 175–184 https://doi.org/10.1016/S0046-8177(87)80336-2 PMID: 3026945
5. Distelmaier F, Calaminus G, and Harms D, et al (2006) Ovarian small cell carcinoma of the hypercalcemic type in children and adolescents Cancer 107 2298–2306
6. McCluggage WG, Oliva E, and Connolly LE, et al (2004) An immuno–histochemical analysis of ovarian small cell carcinoma of hypercalcemic type Int J Gynecol Pathol 23(4) 330–336 https://doi.org/10.1097/01.pgp.0000139644.38835.9d PMID: 15381902
7. Pautier P, Ribrag V, and Duvillard P, et al (2007) Results of a prospective dose-intensive reg- imen in 27 patients with small cell carcinoma of the ovary of the hypercalcemic type Ann Oncol 18 1985–1989
8. Harrison ML, Hoskins P, and du Bois A, et al (2006) Small cell of the ovary, hypercalcemic type—analysis of combined experience and recommendation for management. A GCIG study Gynecol Oncol 100 233–238 https://doi.org/10.1016/j.ygyno.2005.10.024
9. Senekjian EK, Weiser PA, and Talerman A, et al (1989) Vinblastine, cisplatin, cyclophosphamide, bleomycin, doxorubicin, and etoposide in the treatment of small cell carcinoma of the ovary Cancer 64(6) 1183–1187 PMID: 2475239
10. Sholler GL, Luks F, and Mangray S, et al (2005) Advanced small cell carcinoma of the ovary in a pediatric patient with long-term survival and review of the literature J Pediatr Hematol Oncol 27(3) 169–172
11. Callegaro-Filho D, Gershenson DM, and Nick AM, et al (2016) Small cell carcinoma of the ovary-hypercalcemic type (SCCOHT): a review of 47 cases Gynecol Oncol 140(1) 53–57
12. Tewari K, Brewer C, and Cappuccini F, et al (1997) Advanced-stage small cell carcinoma of the ovary in pregnancy: long-term survival after surgical debulking and multiagent chemotherapy Gynecol Oncol 66(3) 531–534 https://doi.org/10.1006/gyno.1997.4801 PMID: 9299272
13. Ulbright TM, Roth LM, and Stehman FB, et al (1987) Poorly differentiated (small cell) carcinoma of the ovary in young women: evidencesupporting a germ cell origin Hum Pathol 18(2) 175–184
14. McCluggage WG, Witkowski L, and Clarke BA, et al (2017) Clinical, morphological and immunohistochemical evidence that small-cell carcinoma of the ovary of hypercalcaemic type (SCCOHT) may be a primitive germ-cell neoplasm Histopathology 70(7) 1147–1154 https://doi.org/10.1111/his.13177 PMID: 28130795
15. Witkowski L, Goudie C, and Ramos P, et al (2016) The influence of clinical and genetic factors on patient outcome in small cell carcinoma of the ovary, hypercalcemic type Gynecol Oncol 141(3) 454–460 https://doi.org/10.1016/j.ygyno.2016.03.013 PMID: 26975901
16. Ramos P, Karnezis AN, and Craig DW, et al (2014) Small cell carcinoma of the ovary, hypercalcemic type, displays frequent inactivating germline and somatic mutations in SMARCA4 Nat Genet 46 427–429 https://doi.org/10.1038/ng.2928 PMID: 24658001 PMCID: 4332808
17. Jelinic P, Mueller JJ, and Olvera B, et al (2014) Recurrent SMARCA4 mutations in small cell carcinoma of the ovary Nat Genet 46 424–426 https://doi.org/10.1038/ng.2922 PMID: 24658004 PMCID: 5699446
18. Witkowski L, Carrot-Zhang J, and Albrecht S, et al (2014) Germline and somatic SMARCA4 mutations characterize small cell carcinoma of the ovary, hypercalcemic type Nat Genet 46 438–443 https://doi.org/10.1038/ng.2931 PMID: 24658002
19. Lin DI, Chudnovsky Y, and Duggan B, et al (2017) Comprehensive genomic profiling reveals inactivating SMARCA4 mutations and low tumor mutational burden in small cell carcinoma of the ovary, hypercalcemic-type Gynecol Oncol 147(3) 626–633 https://doi.org/10.1016/j.ygyno.2017.09.031 PMID: 29102090
20. Chan-Penebre E, Armstrong K, and Drew A, et al (2017) Selective killing of SMARCA2- and SMARCA4-deficient small cell carcinoma of the ovary, hypercalcemic type cells by inhibition of EZH2: in vitro and in vivo preclinical models Mol Cancer Ther 16 850–860 https://doi.org/10.1158/1535-7163.MCT-16-0678 PMID: 28292935
21. Jelinic P, Ricca J, and Van Oudenhove E, et al (2018) Immune-active microenvironment in small cell carcinoma of the ovary, hypercalcemic type: rationale for immune checkpoint blockade J Natl Cancer Inst https://doi.org/10.1093/jnci/djx277 PMID: 29365144
22. Dykgraaf RH, de Jong D, and van Veen M, et al (2009) Clinical management of ovarian small-cell carcinoma of the hypercalcemic type: a proposal for conservative surgery in an advanced stage of disease Int J Gynecol Cancer 19 348–353 https://doi.org/10.1111/IGC.0b013e3181a1a116 PMID: 19407558
23. Woopen H, Sehouli J, and Pietzner K, et al (2012) Clinical experience of young patients with small cell ovarian carcinoma of the hypercalcemic type (OSCCHT) Eur J Obstet Gynecol Reprod Biol 162(5) 313–317