Repurposing Drugs in Oncology (ReDO)—chloroquine and hydroxychloroquine as anti-cancer agents
Ciska Verbaanderd1,2,3, Hannelore Maes2, Marco B Schaaf2, Vikas P Sukhatme4,5, Pan Pantziarka1,6, Vidula Sukhatme4, Patrizia Agostinis2 and Gauthier Bouche1
1Anticancer Fund, Brussels, 1853 Strombeek-Bever, Belgium
2Cell Death Research and Therapy Lab, Department of Cellular and Molecular Medicine, KU Leuven, 3000 Leuven, Belgium
3Clinical Pharmacology and Pharmacotherapy, Department of Pharmaceutical and Pharmacological Sciences, KU Leuven, 3000 Leuven, Belgium
4GlobalCures, Inc, Newton, MA 02459, USA
5Beth Israel Deaconess Medical Center and Harvard Medical School, Boston, MA 02215, USA; Current address: Emory School of Medicine, Atlanta, GA 30322, USA
6The George Pantziarka TP53 Trust, London KT1 2JP, UK
Correspondence to: Gauthier Bouche. Email: gauthier.bouche@anticancerfund.org
Abstract
Chloroquine (CQ) and hydroxychloroquine (HCQ) are well-known 4-aminoquinoline antimalarial agents. Scientific evidence also supports the use of CQ and HCQ in the treatment of cancer. Overall, preclinical studies support CQ and HCQ use in anti-cancer therapy, especially in combination with conventional anti-cancer treatments since they are able to sensitise tumour cells to a variety of drugs, potentiating the therapeutic activity. Thus far, clinical results are mostly in favour of the repurposing of CQ. However, over 30 clinical studies are still evaluating the activity of both CQ and HCQ in different cancer types and in combination with various standard treatments. Interestingly, CQ and HCQ exert effects both on cancer cells and on the tumour microenvironment. In addition to inhibition of the autophagic flux, which is the most studied anti-cancer effect of CQ and HCQ, these drugs affect the Toll-like receptor 9, p53 and CXCR4-CXCL12 pathway in cancer cells. In the tumour stroma, CQ was shown to affect the tumour vasculature, cancer-associated fibroblasts and the immune system. The evidence reviewed in this paper indicates that both CQ and HCQ deserve further clinical investigations in several cancer types. Special attention about the drug (CQ versus HCQ), the dose and the schedule of administration should be taken in the design of new trials.
Keywords: Repurposing Drugs in Oncology (ReDO) project, drug repositioning, chloroquine (CQ), hydroxychloroquine (HCQ), neoplasms, antineoplastic agents, anti-malarial agents.
Copyright: © the authors; licensee ecancermedicalscience. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Published: 23/11/2017; Received: 22/07/2017
Introduction
Chloroquine (CQ) and hydroxychloroquine (HCQ) are both 4-aminoquinoline agents that have been used for more than 70 and 50 years, respectively, to prevent or to treat malarial infections and later also for treating discoid and systemic lupus erythematosus and rheumatoid arthritis. Although HCQ and CQ differ only by one hydroxyl group, the addition of this hydroxyl group results in an important decrease in toxicity, while the efficacy remains constant, at least for malaria [1]. Both drugs are available as generic products and mentioned on the WHO list of essential medicines. Frequently used trade names of CQ include Avloclor, Nivaquine or Aralen, and the most frequently used trade name for HCQ is Plaquenil.
The mechanisms of action of CQ and HCQ against the malarial Plasmodium parasite and against the auto-immune disorders for which they are approved are well known [2–6].
Dosage
The dosage of CQ depends on the indication [3, 4]. It should be noted that CQ is often marketed as chloroquine phosphate (CQ-phosphate) in tablets of 250 mg, which corresponds to about 150 mg of CQ. All doses mentioned below are doses of CQ-phosphate. High doses (1 g of CQ-phosphate per day) are administered in acute phases of malaria or amoebic hepatitis, but only for one or two days. The usual dose for long-term use (rheumatoid arthritis and lupus) is 250 mg of CQ-phosphate per day. For HCQ, doses for long-term use range between 200 and 400 mg per day. Long-term administration of CQ and HCQ in children is not recommended, but doses for long-term treatment between 2 and 5 mg/kg for HCQ have been reported [7].
Toxicity
Short-term administration of CQ or HCQ rarely causes severe side effects. Longer exposure has been associated with some serious though uncommon adverse events [3], including cardiomyopathy [8], irreversible retinal toxicity [9, 10], bone marrow suppression [11] and hypoglycaemia [12]. The risk of retinopathy is increased with large cumulative doses of HCQ (>1000 g). However, daily doses up to 400 mg of HCQ or 250 mg CQ for several years are considered to carry an acceptable risk for CQ-induced retinopathies, with the exception of individuals of short stature [13]. It is advised that patients receiving chronic CQ or HCQ therapy be monitored through regular ophthalmic examinations (3–6 month intervals), full blood counts and blood glucose level checks. CQ has been associated with some cases of diffuse parenchymal lung disease and drug rash with eosinophilia and systemic symptoms (DRESS) syndrome [3]. In case of long-term HCQ exposure, skeletal muscle function and tendon reflexes should be monitored for weakness.
For both CQ and HCQ, specific caution is advised in patients suffering from impaired hepatic function (especially when associated with cirrhosis), porphyria, renal disease, epilepsy, psoriasis, glucose-6-phosphate dehydrogenase deficiency and known hypersensitivity to 4-aminoquinoline compounds [3].
Bioavailability
CQ and HCQ are amphiphilic weak bases with the ability to cross cell membranes easily, which is important for their mechanism of action in malaria treatment and prophylaxis. CQ and HCQ are partially protonated at the physiologic pH (7.4), but they can be trapped in lysosomes (pH 4–5) because of bi-protonation [14]. CQ has pKa values of 8.4 and 10.2, while HCQ has pKa values of 8.3 and 9.7 [14, 15].
Both CQ and HCQ have a high bioavailability, 89% and 74% respectively, and a large distribution volume after oral administration. Roughly, 50%–70% of CQ is protein-bound in the plasma [3]. The terminal elimination half-life of CQ is 1–2 months and for HCQ approximately 50 days in blood (32 days in plasma). Both drugs are partially metabolised by hepatic dealkylation, but they differ in the number of the metabolites produced. The active metabolites of CQ are monodesethylchloroquine and bisdesethylchloroquine, while HCQ has one extra active metabolite, namely desethylhydroxychloroquine. Moreover, CQ and HCQ are slowly excreted and may still be detected in urine several months after administration [3, 4, 9].
Pre-clinical evidence in cancer—in vivo
CQ and HCQ have been extensively studied both in vitro and in vivo in various cancer types. This paper focuses on the results from in vivo research, since this is most relevant to clinical practice. Both drugs can be administered as monotherapy or as adjuvant agents to increase the efficacy and to limit drug resistance of standard anti-cancer therapy.
Monotherapy
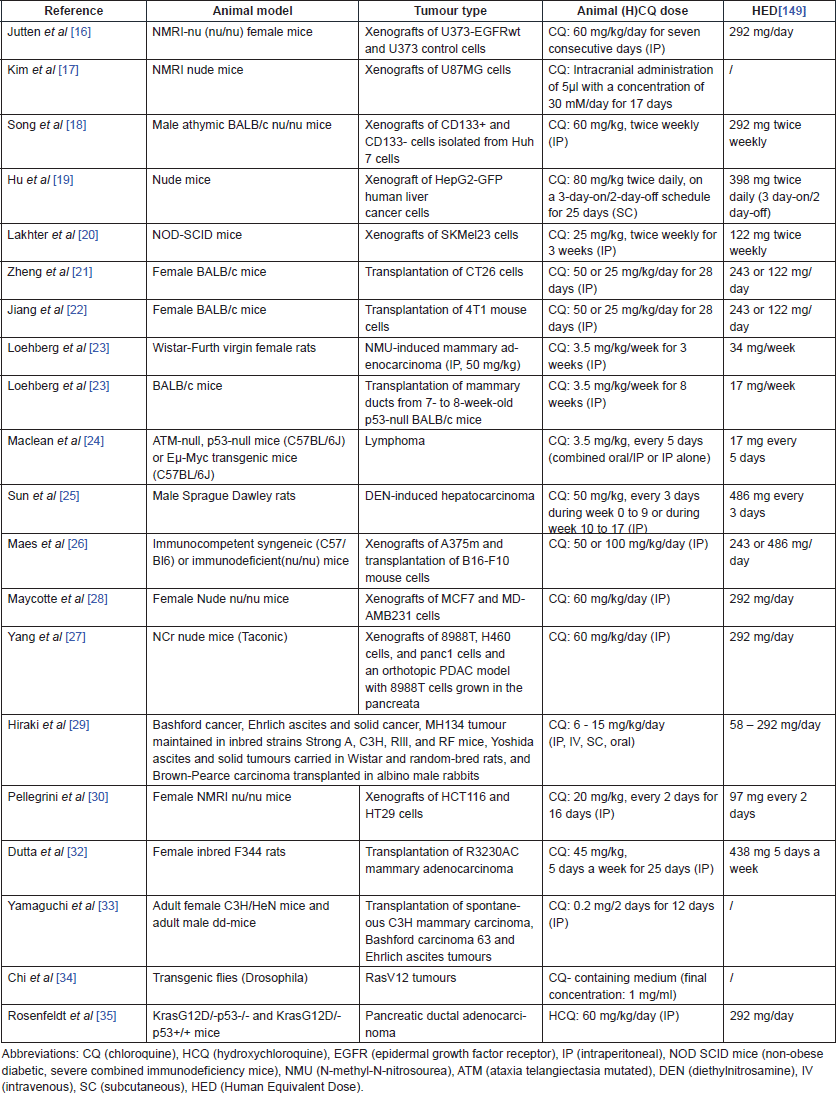
Table 1 lists the main characteristics [animal models, tumour types, animal (H)CQ doses and human equivalent doses (HED)] of the in vivo studies performed with CQ or HCQ alone.
Table 1. In vivo studies investigating the efficacy of CQ and HCQ monotherapy.
Starting with in vivo studies that observed beneficial effects of CQ administration in cancer, Jutten et al noted a delayed tumour growth in mice bearing epidermal growth factor receptor (EGFR)-overexpressing glioblastoma xenografts in response to CQ administration. In addition, the time to reach four times the initial tumour volume was significantly longer in the CQ-treated group [16]. Kim et al confirmed this observation in another glioblastoma xenograft mouse study, where CQ was injected intracranially. They observed that the number of mitotic cells was significantly reduced and the number of apoptotic cells was increased after CQ administration [17]. In addition, a significant reduction of tumour volume and tumour incidence was shown by Song et al [18] in mice bearing liver cancer stem cells and Hu et al [19] observed significant tumour growth and weight reduction in an orthotopic xenograft model of liver cancer after CQ administration. Lakhter et al [20] demonstrated that CQ significantly reduced both tumour volume and tumour mass in a human melanoma xenograft model. Zheng et al [21] showed reduced tumour progression and prolonged survival time (not significant) in colon cancer-bearing mice when administering either 25 or 50 mg/kg of CQ.
Doses of 25 and 50 mg/kg of CQ both significantly increased survival time and reduced primary tumour volume in mice implanted with a highly metastasizing breast cancer cell line, as shown by Jiang et al. Interestingly, the number and diameter of lung metastases was reduced as well, and CQ enhanced tumour cell apoptosis in the high dose group [22].
The incidence of mammary tumours and their growth rate was significantly lower and tumour onset was delayed in CQ-pre-treated rats after being subjected to mammary adenocarcinoma induction using N-methyl-N-nitrosourea (NMU), as shown by Loehberg et al. In wild-type BALB/c mice transplanted with mammary ducts of BALB/c p53-null mice, CQ pre-treatment did not affect tumour incidence [23]. Maclean et al confirmed that CQ could not prevent spontaneous tumour formation in p53-deficient mice. In contrast, intermittent CQ administration significantly reduced the tumour development and doubled the overall survival (OS) of Eμ-Myc mice [24].
Furthermore, Sun et al showed that CQ administration is effective in reducing tumour growth in rats with established hepatocarcinoma. In contrast, CQ promoted tumour development in the earlier so-called dysplastic stage, clearly illustrating the dual role of autophagy in tumour formation (see section on mechanisms of action) [25]. Finally, Maes et al [26] reported that either a dose of 50 mg/kg or a dose of 100 mg/kg of CQ can reduce tumour growth and cell proliferation, dependent on the cell type. Of note, this study showed that CQ not only inhibits autophagy but also affects the tumour microenvironment and tumour vasculature. The exact working mechanisms will be clarified in the section on mechanisms of action.
Some studies noted that the efficacy of CQ application in anti-cancer therapy depends on the tumour type that is being treated and suggested that the autophagy dependency of tumour cells might play a role [27, 28]. For example, tumour growth was significantly reduced in an MDAMB231 xenograft mouse model, but not in an MCF-7 xenograft mouse model, while both models showed signs of autophagy inhibition after CQ treatment [28]. A similar observation can be made when comparing CQ efficacy in pancreatic cancer mouse models and a lung cancer mouse model: CQ significantly slowed down tumour growth and increased survival in the first, but not in the latter [27]. Another study by Hiraki et al [29] investigated the effects of CQ in various in vivo cancer models and demonstrated that CQ is more effective in connective tissue-rich Bashford and Brown–Pearce tumours than in Ehrlich, Yoshida and MH134 tumours.
A lack of efficacy in certain tumour models could potentially be explained by a study performed by Pellegrini et al exploring the effects of CQ under acidic conditions, which mimics the tumour environment. CQ bi-protonation under those conditions could impede cytotoxicity, because the cellular uptake of CQ is reduced. This observation highlights a possible limitation of CQ in anti-cancer therapy. However, the sensitivity of tumour cells might be restored using tumour pH-modulating agents [30]. Ironically, hypoxic cells that can increase acidification of the extracellular space through anaerobe glycolysis are often more autophagy-dependent and, therefore, more sensitive to CQ treatment, as shown by in vitro studies [31].
A limited amount of studies reported potential detrimental effects of CQ and HCQ in in vivo cancer models. First, CQ pre-treatment of rats one week before a subcutaneous injection with mammary adenocarcinoma and follow-up treatment for 18 days following this event significantly enhanced tumour weight and volume in these rats [32]. Second, in a 1966 paper, very low CQ doses (0.2 mg/2 days) led to a more infiltrative morphological pattern of the advancing margin of subcutaneously transplanted mammary carcinoma in mice [33]. Third, malignant tumour growth and metastasis of Ras(V12) cells is observed in transgenic drosophila models after CQ administration [34]. Importantly, HCQ promoted tumour growth in Ras-driven pancreatic tumours developing without p53(KrasG12D/ p53–/–) [35]. Collectively, these results on a possible detrimental effect of (H)CQ emphasise the importance of the specific tumour setting and tumour characteristics when targeting autophagy (see section on mechanisms of action) [35, 36].
Combination therapy
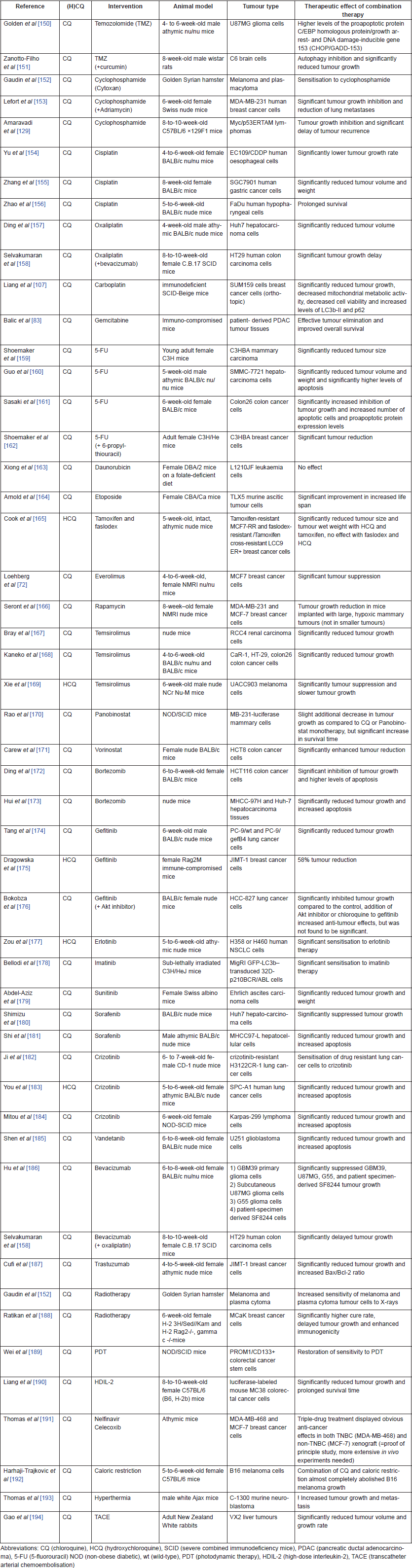
Table 2 summarises the information from articles that studied the effect of CQ (n = 46) or HCQ (n = 5) in vivo in combination with other therapies. A more interesting and still under-explored treatment approach for a complex disease such as cancer is to combine various anti-cancer agents acting at different levels in the tumour cells and microenvironment [37]. Interestingly, CQ and HCQ have already been tested in combination with over 40 other drugs in preclinical cancer research. Both CQ and HCQ can effectively increase the efficacy of various anti-cancer drugs, which is further explained in the section on mechanisms of action. Therapies used in combination with CQ or HCQ include chemotherapeutic drugs, tyrosine kinase inhibitors, various monoclonal antibodies, hormone therapies and radiotherapy (Table 2).
Table 2. Overview of in vivo research combining known anti-cancer agents with either CQ or HCQ.
Human data
Numerous clinical trials in which either CQ or HCQ is being used to treat patients with a range of cancer types are registered in clinical trial databases. In clinical trials, these drugs are most often administered in combination with other anti-cancer agents. More information on the registered clinical trials is provided in Tables 3 and 4, for CQ and HCQ, respectively. Few trials have been completed. Therefore, limited published data are available on the safety and therapeutic efficacy of these antimalarial drugs in cancer. A schematic overview of the published clinical trial data of CQ and HCQ can be found in Tables 5 and 6, respectively.
In the next section, the clinical effects of CQ and HCQ will be discussed separately because important differences can be observed in toxicity and efficacy of both drugs.
Table 3. Information on clinical trials investigating CQ use in cancer (Source: ClinicalTrials.gov).
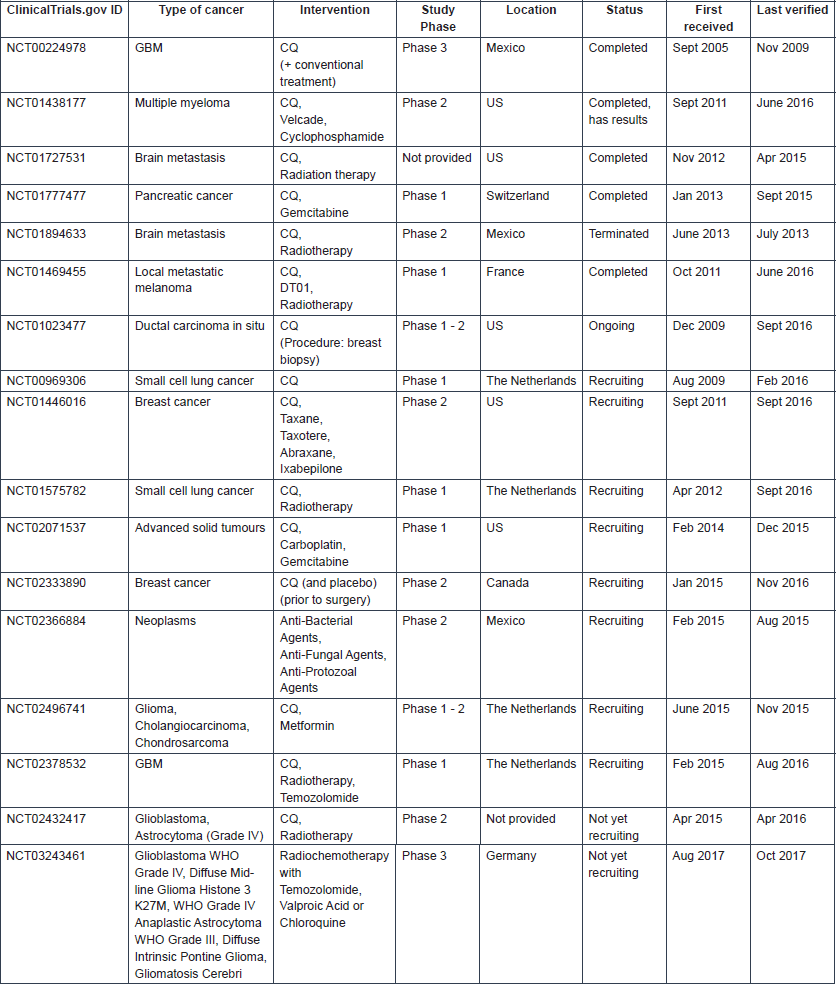
Table 4. Information on clinical trials investigating HCQ use in cancer (Source: ClinicalTrials.gov).
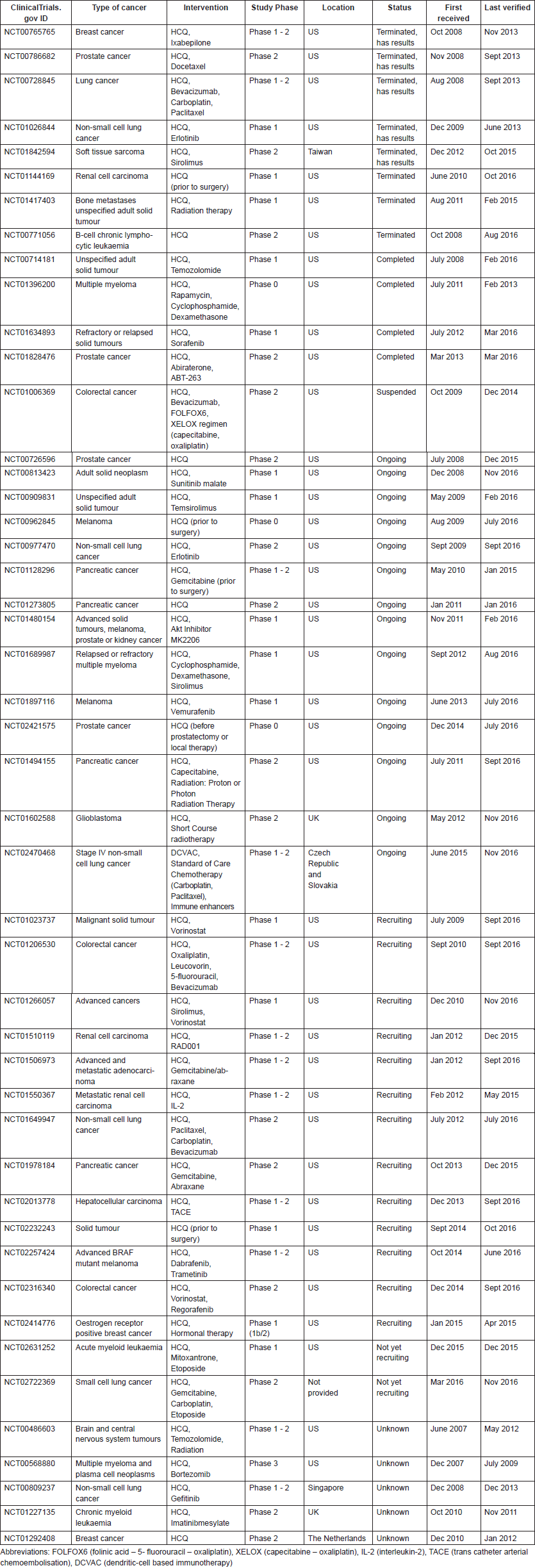
Table 5. Publications reporting clinical trial results on CQ use in cancer.
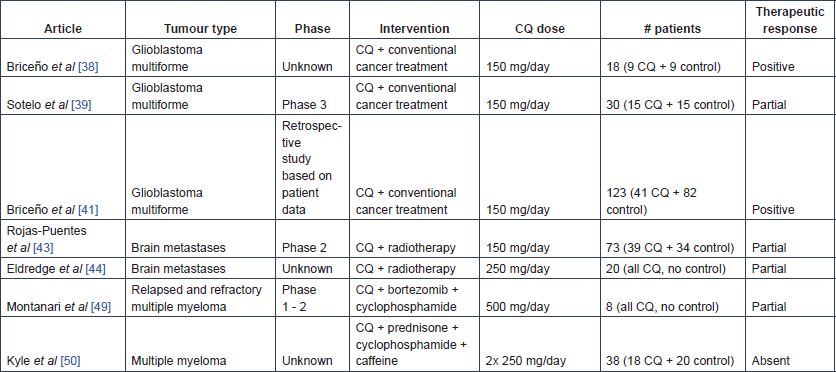
Table 6. Publications reporting clinical trial results on HCQ use in cancer.
CQ
Glioma and brain metastases
In May 1998, one of the first clinical trials on CQ use in cancer was started, which was an open, prospective, randomised controlled study with 18 glioblastoma multiforme (GBM) patients [38]. The test group consisted of nine patients who received 150 mg CQ daily after resection of the lesion, in addition to radiotherapy (total dose of 6000 Gy) and four cycles of carmustine-chemotherapy every six weeks (200 mg/m2), while the nine patients in the control group received placebo instead of CQ. In the abstract of this study, the authors reported that adjuvant CQ administration significantly enhanced patient survival [33 ± 5 months for CQ-treated patients and 11 ± 2 months for controls (p < 0.0002)]. Due to some inconsistencies in the report, the calculation of the mean survival in the CQ-treated group is unclear, but the Kaplan Meier analysis remains significant. A higher seizure frequency was observed in the CQ-treated group and could not be explained. However, standard antiepileptic treatment was reported to easily suppress these seizures. The same group of researchers started a similar randomised, double blind, placebo-controlled study in October 2000 [39]. In this second study, 15 GBM patients received 150 mg CQ each day for 12 months after surgery in combination with their conventional anti-cancer therapy, four cycles of carmustine-chemotherapy every five weeks (200 mg/m2) and a total radiation dose of 60 Gy; the other 15 patients received adjuvant placebo treatment. A median survival time of 24 months was observed in the CQ-treated group, as compared with 11 months in the control group. In addition, the hazard ratio for death was approximately half as large in the patients receiving CQ though this was not statistically significant (hazard ratio: 0.52, [95% CI 0.21–1.26, p = 0.139]). No important adverse effects were noted in this trial. The small sample size is an important limitation in both studies, and larger clinical trials are needed to confirm the efficacy of CQ in GBM therapy [39, 40]. In a retrospective study, the same research group looked at data collected over five years from 41 GBM patients in Mexico who received adjuvant CQ therapy and did not participate in the previously mentioned clinical trials [41]. The mean survival time of these CQ-treated patients was significantly longer compared with a control group of 82 glioblastoma patients [25 ± 3.4 months and 11.4 ± 1.3 months after surgery respectively (p = 0.000)].
After the observation of promising outcomes in five recurrent GBM patients treated with 250 mg CQ a day and reirradiation for 20 months [42], a phase-2 clinical trial tested the effects of CQ as a radio-sensitising agent in patients with brain metastases [43]. In this trial, 39 patients were administered whole-brain irradiation (30 Gy in 10 fractions over two weeks) in combination with a daily dose of 150 mg CQ for four weeks, while 34 patients received placebo instead of CQ in addition to the same radiation treatment. The overall response rate or OS did not improve after CQ administration. However, the progression-free survival of brain metastases rate was increased (CQ-treated group: 83.9% [95% CI 69.4–98.4] and control group: 55.1% [95% CI 33.6–77.6] (at one year), relative risk: 0.31 [95% CI 0.1–0.9, p = 0.046]). The absence of adverse effects and the improved local control of brain metastases indicate that CQ might be a useful addition to whole brain irradiation in patients with brain metastases. In a prospective, single-cohort study of 20 patients with brain metastases from solid tumours, 250 mg CQ daily was administered for five weeks in combination with whole-brain irradiation [44]. The intracranial response rate corresponded to an objective clinical response of 93% after three months of whole-brain irradiation, there was a slight, positive trend in OS (median OS of 5.7 months, compared with 4.2 months for patients in class II estimated by the radiation therapy oncology group recursive partitioning analysis), and no adverse reactions were detected. Finally, two case reports mentioned unusual skin reactions after concomitant use of CQ and radiation, illustrating the radio-sensitising effect of CQ [45, 46].
In a paediatric patient with a recurrent BRAF V600E mutant brainstem ganglioglioma, tumour growth was blocked and vemurafenib sensitivity restored following treatment with 150 mg CQ daily for at least 30 months [47, 48]. The same research group reported in vitro and ex vivo data showing that autophagy inhibition was able to improve the response to BRAF inhibition in resistant tumour cells [48]. Next, CQ was administered to two patients with acquired resistance to BRAF inhibition. The first patient was treated with standard doses of vemurafenib plus 250 mg daily of CQ during focal radiation of large primary lesions. Vemurafenib was continued and the CQ dose was increased to 500 mg daily after completion of radiation. A rapid favourable clinical response to the combination therapy was observed in as little as six weeks and was maintained for seven months, at which point the patient had to stop therapy for unrelated medical issues. The second patient was treated with 500 mg CQ daily in combination with standard dosing of vemurafenib. Acquired resistance to vemurafenib was overcome within four weeks of the addition of CQ and clinical improvement could be observed, which was maintained for two and a half months. However, therapy had to be stopped and the family chose to pursue palliative therapy afterwards.
Multiple myeloma
Eleven patients with relapsed and refractory multiple myeloma were enrolled and treated with 500 mg CQ daily (on days 1–14 and 22–35) in addition to bortezomib and cyclophosphamide (administered orally twice daily) in a phase-2 clinical trial [49]. Of these eleven patients, only eight patients were evaluable. CQ was able to partially restore the bortezomib sensitivity: three patients had a partial response, one had stable disease and four had progression as best responses.
The adjuvant therapeutic effect of 250 mg CQ twice daily in combination with cyclophosphamide and prednisone was tested for a period of ten days in 38 myeloma patients [50]. Twenty patients received cyclophosphamide and prednisone, while the other 18 patients received extra treatment with CQ and caffeine, but no additional response was observed in the CQ-treated patient group.
HCQ
Solid cancers
The effect of HCQ and temsirolimus combination therapy was tested in 27 patients with advanced solid cancer during a phase-1 dose-escalating study and subsequently in 13 patients with metastatic melanoma at the phase-2 recommended dose [51]. No patient experienced an objective response but 19 out of the 27 phase-1 patients (73%) and 9 out of 13 melanoma patients (69%) had stable disease. In patients with stable disease, HCQ addition was shown to produce metabolic stress in the tumours. Inhibition of autophagy (see section on mechanisms of action), measured by counting the number of autophagic vacuoles per cell in tumour tissues and peripheral blood mononuclear cells of patients, was only noted in patients receiving at least 1200 mg HCQ daily. This study recommends an adjuvant HCQ dose of 600 mg, twice daily.
Next, the combinatory effect of HCQ and temozolomide was investigated in 40 cancer patients with advanced solid tumours and melanoma, and the recommended dose of 600 mg twice daily was confirmed [52]. HCQ was shown to successfully inhibit autophagy, as evidenced by the significant accumulation of autophagic vacuoles in peripheral blood mononuclear cells (mean autophagic vacuole counts: 2.19 at baseline, 2.45 after HCQ treatment, 3.84 after treatment with HCQ plus TMZ [difference between HCQ plus TMZ and baseline: p = 0.0007, difference between HCQ plus TMZ and HCQ only: p = 0.0034]).
The safety and preliminary efficacy of HCQ and vorinostat combination treatment was tested during a phase-1 study in 27 patients with advanced solid tumours [53]. In this study, the maximum-tolerated HCQ dose was set at 600 mg daily in combination with 400-mg vorinostat. A confirmed durable partial response was observed in a renal cell carcinoma patient, and prolonged stable disease was seen in two colorectal cancer patients. In contrast to the previous study, autophagy was not significantly affected.
Finally, in a pilot retrospective study, 25 stage-IV cancer patients (various types) who had no clinical response to maximally tolerated chemotherapy and to first-line metronomic chemotherapy were treated with sirolimus (2 mg/day) and the autophagy inhibitor HCQ (400 mg/day) in addition to their current metronomic chemotherapy for at least three months. The therapy was reported to be relatively safe, and the overall response rate was 40%, with an 84% disease control rate [54]. However, this was a retrospective analysis requiring cautious interpretation.
Glioblastoma
The efficacy and safety of HCQ was studied in combination with radiotherapy and temozolomide in 92 GBM patients during a phase-1–2 study [55]. This study indicated a maximum tolerated dose (MTD) of 600 mg HCQ a day in this therapeutic setting. OS did not seem to be affected in comparison with the temozolomide arm of the trial reported by Stupp [56], and autophagy was not found to be consistently inhibited in all patients.
Lung cancer
The combination of HCQ with erlotinib can be used safely in daily doses of 150 mg erlotinib and 1000 mg HCQ, as determined by a phase-1 study in 27 patients with advanced non-small cell lung cancer (NSCLC) [57]. Of the 19 patients who remained in the study, one had a partial response and four had stable disease as best response. Subsequent ophthalmic surveillance on seven trial participants who had taken HCQ for a duration longer than six months showed that retinal toxicity occurred in two patients after 11 and 17 months of exposure [58].
This highlights the importance of retinal toxicity monitoring (via high-resolution spectral-domain optical coherence tomography, fundus auto fluorescence imaging, Humphrey visual field testing and multifocal electroretinography) during clinical trials with HCQ.
Multiple myeloma
During a phase-1 study, the safety of the combination of HCQ and bortezomib was explored in 25 patients with relapsed or refractory myeloma [59]. A dose of 600 mg HCQ twice daily was reported to be safe and tolerable in combination with standard doses of bortezomib. The increase in the number of autophagic vacuoles was not significantly associated with clinical response or HCQ exposure. Of 22 evaluable patients, three (14%) had very good partial responses, three (14%) had minor responses, ten (45%) had stable disease for at least one cycle and six (27%) had immediate progression.
Pancreatic cancer
The safety of the combination of pre-operative HCQ (1200 mg daily) and gemcitabine administration was demonstrated in 35 patients with pancreatic adenocarcinoma in a phase-1–2 trial [60]. This study reported promising clinical response markers (e.g. CA 19–9 biomarker and R0 resection rate). An exploratory analysis showed significantly improved disease-free survival and OS (15.03 versus 6.9 months and 34.83 versus 10.83 months, respectively) in patients for whom autophagy was sufficiently inhibited (n = 8) (at least 51% increase in the autophagy marker LC3B-II in peripheral blood mononuclear cells) compared with other patients (n = 9).
Next, a phase-2 study investigated the safety and efficacy of HCQ monotherapy with either 400 or 600 mg two times a day in 20 patients with previously treated metastatic pancreatic cancer, but no significant differences were observed between groups [61]. In addition, inhibition of autophagy could not be achieved consistently, as shown by LC3B-II analysis in the lymphocytes of patients, and the two-month progression-free survival rate was only 10%.
Sarcoma
The combination of 1 mg sirolimus and 200 mg HCQ twice daily for two weeks was tested in ten sarcoma patients who had failed first-line treatment [62]. This study started from the hypothesis that there is metabolic symbiotic relationship between cancer-associated fibroblasts (CAFs) and sarcoma cells (see mechanisms of action). The study showed that this relationship might be altered by treatment with sirolimus and HCQ as glycolysis was inhibited within the tumours. Based on FDG PET response criteria, two weeks after treatment initiation, six patients showed partial response, three had stable disease and one had progressive disease. However, most patients discontinued treatment before the initially planned eight-week response assessment, for disease progression.
Mechanism of action
Multiple hypotheses have been proposed on how CQ and HCQ exert their anti-cancer activity. Most studies reported the direct action of these drugs on cancer cells, but more recent studies have also mentioned important effects of CQ and HCQ on the tumour microenvironment. Based on preclinical studies, it is safe to say that CQ and HCQ have multiple mechanisms of action that might complement each other.
The most relevant and evidence-based mechanisms of action of CQ and HCQ in anti-cancer treatment will be briefly explained in the next section. The benefits of combining these antimalarial drugs with existing anti-cancer treatments will also be described. In the final section, the variation in sensitivity of cancer patients to CQ and HCQ therapy will be clarified.
Direct anti-tumour effects
The main and most studied anti-cancer effect of CQ and HCQ is the inhibition of autophagy, but other preclinically proven anti-cancer activities of the antimalarial agents include influencing the TLR9/nuclear factor kappa B (NF-κB) signalling pathway, the CXCL12/CXCR4 signalling pathway and the p53 pathway.
Autophagy inhibition
Autophagy literally means ‘self-eating’. It is a process in which a cell destroys old or defective cellular components, thereby releasing cellular building blocks including nucleotides, amino acids and fatty acids. Those degradation products can later be recycled by the cell to meet its metabolic needs. Autophagy is an essential intracellular process to ensure cell survival under stressful conditions (e.g. hypoxia, starvation and organelle damage). Different types of autophagy exist but, here, we will use the term to refer to macroautophagy. Autophagy is a complex multi-faceted process [63]. One putative biomarker is the level of LC3B-II, an essential protein during autophagosome formation and the level of scaffolding protein p62 [or sequestosome 1 (SQSTM1)] [64, 65]. Autophagy has both pro-tumour and anti-tumour functions, which may be both stage and tissue-type specific.
In early carcinogenesis, autophagy has a tumour suppressive role since it has an important quality control function and protects the cell by sequestering and eliminating defective cellular components, such as damaged mitochondria, and by maintaining cellular homeostasis [66, 67]. In addition, several autophagic proteins can directly suppress tumour formation (e.g. Beclin-1, UVRAG and Bif-1) and autophagy has been shown to degrade tumour promoting proteins as well (e.g. p62/SQSTM1) [68]. In line, deregulation of autophagy has been repeatedly associated with human cancers [67].
In contrast, autophagy can promote tumour growth in more advanced stages of cancer [69]. Pro-survival autophagy is induced in response to a variety of stressful conditions including but not limited to, starvation, loss of proteostasis, organelle damage and hypoxia. Some anti-cancer treatments can also induce pro-survival autophagy. Autophagic properties such as nutrient recycling can support cancer cell survival. Moreover, key regulators of cell growth can be degraded and the DNA damage response can be suppressed through increased autophagy [66–68]. Therefore, inhibition of autophagy can be an interesting anti-cancer strategy when cancer cells start depending on autophagy for survival, a moment called the autophagic switch [64, 70].
CQ and HCQ inhibit the autophagic flux at a late stage (Figure 1): the fusion of the autophagosomes with the lysosomes and subsequent degradation of the autolysosome. Upon entering the lysosomes, CQ and HQ become protonated, which leads to their entrapment in acidic lysosomes and an increase in the lysosomal pH, which inhibits the lysosomal degradative enzymes [71]. Loehberg et al [72] suggested that CQ might also modulate autophagy by modifying the PI3K/Akt/mTOR pathway.
In summary, autophagy plays a dual role in cancer and the success of autophagy inhibition, using the late stage inhibitors CQ and HCQ, depends on the timing and context. Autophagy is an interesting therapeutic target after the autophagic switch. However, the autophagy dependency of the tumour cells and any combinatory therapies can influence the sensitivity to autophagy inhibition, which will be discussed later.
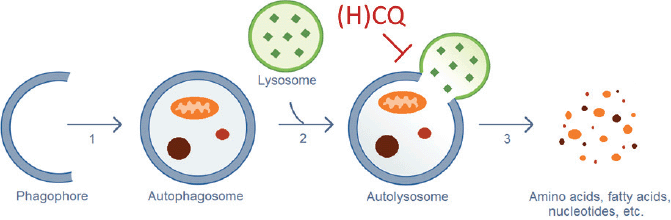
Figure 1. Autophagic process. (1) Elongation of the phagophore and vesicle formation. (2) Fusion of the autophagosome and a lysosome. (3) Destruction of the engulfed cellular components by lysosomal hydrolases. CQ and HCQ inhibit autophagy through interference with the lysosomal acidification (Step 2).
Inhibition of the TLR9/nuclear factor kappa B signalling pathway
TLR9, a member of the Toll-like receptor family, is located in the endosomal compartment. This receptor recognises unmethylated single stranded DNA and is necessary for pathogen recognition and innate immune system activation. In cancer, expression and stimulation of TLR9 is linked with invasiveness, as shown in in vitro experiments [73–75]. Moreover, the expression levels of TLR9 are higher in hepatocellular carcinoma, oesophageal, lung, breast, gastric and prostate cancer cells as compared with adjacent noncancerous cells, and high expression is often linked with poor prognosis [73–76]. Because of this observation, it was suggested that TLR9 might be an appropriate anti-cancer target [73, 74, 76].
The TLR9-mediated activation of the NF-κB signalling pathway and the associated enhanced expression of matrix metalloproteinase-2 (MMP-2), MMP-7 and cyclo-oxygenase 2 mRNA, all factors associated with tumour progression and migration, can explain the role of TLR9 in cancer [73, 74]. At first, CQ was thought to inhibit this pathway by inhibiting endosomal acidification. However, CQ most likely modifies the structure of the nucleic acids responsible for TLR activation to prevent binding to TLRs [77]. An in vitro study also showed that invasion of brain cancer cells is hypoxia-induced through upregulation of TLR9 expression, which could be significantly inhibited by CQ [78].
In contrast, low expression of TLR9 is reported to be associated with a poorer prognosis in patients with triple-negative breast cancer. CQ had a promising effect on tumour growth and invasiveness, independent of the TLR9 status in triple-negative breast cancer cells in vitro, but it did not reduce the growth of orthotopic triple-negative breast cancer tumours in vivo [79, 80].
Inhibition of CXCL12/CXCR4 signalling
The interaction between the CXCR4 chemokine receptor and its ligand CXCL12 plays a major role in chemotaxis and adhesion of cells, and secretion of growth factors. In recent years, research has shown an association between CXCL12/CXCR4 signalling and cancer progression [81, 82]. This interaction is said to influence the invasive phenotype of pancreatic cancer for example.
In 2012, a CXCR4 small molecule antagonist (NSC56612), structurally resembling CQ and HCQ, was identified through in silico modelling of this receptor [82]. Next, CQ and HCQ were tested via in vitro assays, in which they were found to suppress pancreatic cancer cell proliferation [82, 83]. Mechanistic studies have shown that CQ, at least, partially inhibits CXCL12/CXCR4 signalling, as demonstrated via reduced phosphorylation of the extracellular signal-regulated kinase (ERK) and the signal transducer and activator of transcription 3 (STAT3). Interestingly, CQ and HCQ can induce CXCR4 internalisation in cancer stem cells, making these cells less sensitive to CXCL12 signals [83].
Furthermore, a study in a pancreatic cancer patient-derived xenograft model showed that CQ specifically targets highly aggressive cancer stem cells through inhibition of their self-renewal process. Thus, CQ could be useful to block cancer stem cell-metastasis and may be combined with other anti-cancer agents (e.g. gemcitabine) that target the bulk of the tumour [83].
Interference with the p53 pathway
The tumour suppressor protein p53 plays an essential role in maintaining an error-free genome and inducing cell death in case the damage is irretrievable. Therefore, it is a key protein in the prevention of tumour development [84].
Both in vitro and in vivo research has indicated that CQ can stabilise the p53 protein and activate the p53-dependent transcription of pro-apoptotic genes [17, 23, 24, 72, 84, 85]. Several hypotheses have been proposed to explain the underlying mechanism, but there is no definite answer yet. One of these hypotheses is that CQ intercalates in DNA, which leads to structural changes and thus induction of p53 [17, 85]. Moreover, the p53 activation by CQ might be mediated by the ataxia telangiectasia mutated protein, dependent on the cell type [17, 23, 24]
There is also some discussion about the relationship between the p53 status and the effects of autophagy inhibition on cancer development. Several studies report an accelerated tumour development when autophagy is inhibited in mice without p53 [35, 86, 87], but Yang et al [88] showed that that inhibition of autophagy could still have beneficial effect in p53 mutant tumours. Other studies confirmed that CQ exerts anti-cancer effects independent of the p53 pathway and the p53 status [88–90]. Synergy between the p53-dependent and -independent mechanisms of CQ is likely [17].
Recently, a p53-dependent mechanism was reported in which CQ induces tumour suppressor protein Par-4 secretion, triggering paracrine apoptosis of cancer cells and inhibition of tumour metastasis. This mechanism involves the CQ-dependent activation of p53 and the subsequent induction of Rab8b, which is necessary for transport of vesicles of Par-4 to the plasma membrane [91].
Moreover, CQ might prevent degradation of a p53-related protein, called Bcl homology-3-only protein p53 upregulated modulator of apoptosis (PUMA), as shown in mice studies. CQ increased the levels of PUMA, without affecting p53 in these studies [20, 92].
Other potential mechanisms of action
Additional mechanisms have been suggested, but they have not been studied to the same extent and will only be briefly described here.
In recent years, it has become clear that glutaminolysis plays an important role in metabolic processes associated with cancer cell proliferation and survival. Therefore, targeting glutaminolysis could provide novel approaches to improve cancer treatment [93]. It was shown that CQ affects glutamate dehydrogenase activity [94–96], which could be a potential mechanism of action in anti-cancer treatment. The hypothesis of inhibiting metabolic processes using metformin and CQ is currently being tested in one clinical trial with patients with isocitrate dehydrogenase 1 and 2 (IDH1/2)-mutated chondrosarcoma, glioma and intrahepatic cholangiocarcinoma [97].
CQ and HCQ can activate caspase-3 and modulate the Bcl-2/Bax ratio inducing apoptosis in CLL, B-cell CLL and glioblastoma cells [17, 89, 98–100]. CQ-mediated cell-cycle-arrest and apoptosis was observed in breast cancer cells and was associated with a decrease in protein levels/activity of polo-like kinase 1 (Plk-1), ERK1/2 Akt and cell division cycle 25C (CDC25C). The same study described induction of caspase-3-mediated spindle abnormalities and down regulation of the mitochondrial transmembrane potential by CQ [101]. A decreased lung cancer cell growth after low CQ concentrations was ascribed to an increased lysosomal volume and a phosphatidylcholine-specific phospholipase C involvement (PC-PLC). Higher CQ concentrations still induce apoptosis and necrosis, but likely via different processes [102].
Moreover, HCQ might affect acetylation status in the N-terminal lysines of histones H3 and H4, thus modulating cell growth and differentiation, as shown in human breast cancer cells [103].
In addition, CQ might directly affect Hedgehog signalling. Under normal conditions, this is a quiescent pathway, but activation can cause tumorigenesis and maintains cancer stem cells. Anti-cancer treatment options targeting this specific pathway have been explored, but this has yielded little results so far [104]. One study suggested that CQ might modulate protein levels of the Hedgehog signalling pathway (smoothened, patched and GLI1 proteins) [83].
CQ can inhibit hypoxia-stimulated metastasis via modulation of hypoxia-inducible factor 1α (HIF-1α), vascular endothelial growth factor (VEGF), and epithelial mesenchymal transition (EMT) as shown in a cholangiocarcinoma cell line [105].
In triple-negative breast cancer, CQ was shown to eliminate cancer stem cells through reduction of the expression of Janus-activated kinase 2 and DNA methyl transferase 1 [106] or through induction of mitochondrial dysfunction, subsequently causing oxidative DNA damage and impaired repair of double-stranded DNA breaks [107].
Of note, various studies showed growth inhibition of melanoma cells after CQ administration, but this inhibition was more pronounced in pigmented melanoma, which could be ascribed to CQ’s high affinity for melanin [108]. There is also some contradictory evidence about a potential link between Burkitt’s lymphoma incidence and CQ administration [109, 110].
Modulation of tumour micro-environment
Immunomodulation
An increasing level of research is addressing the essential role of the immune system in cancer development. Activating the immune system against cancer cells is becoming a promising therapeutic approach [111], as immune cells have the ability to detect and destroy malignant cells [66].
Interestingly, autophagy and lysosomal function have been found to be involved in both innate and adaptive immunity [66]. Therefore, inhibitors of these processes such as CQ and HCQ could potentially modulate the immune system and subsequently influence tumour development. However, lysosomal function and autophagy have a dual role in the anti-tumour immune response. Activation of these processes could both activate and impair the immune response, dependent on the circumstances [66]. In addition, autophagy and lysosomal function affect the response of tumour cells to the immune system as well. For example, tumour cell autophagy can generate mediators that provoke an immune response via modulation of the tumour cell secretome and surface proteome, but it may also help tumour cells to escape the immune system [64, 66].
In general, the interaction between cancer cells and the immune system is complex and further research is warranted to determine when CQ or HCQ administration can lead to beneficial effects in the context of anti-tumour immunity [112]. This is particularly important if CQ or HCQ would be considered for use in combination with immunomodulation anti-cancer therapies.
Normalisation of the tumour vasculature
The tumour vasculature, responsible for supplying the tumour with nutrients and oxygen, is an important component in the tumour micro-environment and plays an essential role in tumour cell metastasis [113]. A first therapeutic approach is to destroy blood vessels in order to block the nutrient and oxygen supply to the tumour. However, accumulating evidence suggests that improving the highly abnormal tumour vessel structure, also called vessel normalisation, is preferred over anti-angiogenic therapy. The benefits of vessel normalisation include a decrease in tumour hypoxia, reduced cancer cell intravasation and metastasis, and an increase in chemotherapeutic drug delivery and response [114, 115].
A recent study showed that CQ normalises tumour vessels, independent of its autophagy inhibitory effect, through reduction of vessel density and improvement of cell alignment and formation of tight junctions. At the molecular level, CQ alters endosomal Notch1 trafficking and signalling in endothelial cells, hereby increasing the quiescent phenotype of the endothelial cells [26, 116]. Of note, systemic CQ administration has also been shown to reduce the vascular toxicity of the intratumorally administered, anti-tumour agent Transferrin-CRM107 in in vivo glioma models [117].
Disruption of the CAF—cancer cell interplay
The final interplay between the tumour and its microenvironment that may be influenced by CQ involves CAFs [118]. Glutamine and caveolin-1 are key players in this autophagy-mediated interplay, in which CAFs and tumour cells support each other through glutamine production/secretion and autophagy stimulation. Interestingly, this interplay can be uncoupled through the autophagy inhibitory or lysosomotropic activity of CQ, but the exact mechanism should still be clarified [119, 120].
Synergism with approved anti-cancer drugs
Existing anti-cancer therapies often induce pro-survival autophagy in cancer cells, which is associated with therapeutic resistance. Because of their ability to inhibit autophagy, CQ and HCQ are able to sensitise tumour cells to chemotherapy and radiation. Therefore, these drugs are often tested in (pre)clinical research in combination with other anti-cancer therapies. Though, some caution is advised when concomitantly using these antimalarial drugs with other anti-cancer agents because autophagy can also be inhibited in normal cells, which causes unwanted toxicity (e.g. nephrotoxicity) [121].
CQ-mediated sensitisation to anti-cancer therapy has also been ascribed to autophagy-independent mechanisms. As mentioned earlier, chemotherapeutics can reach the tumour site more easily after tumour vessel normalisation [26, 116]. Moreover, CQ can prevent the entrapment of protonated chemotherapeutic drugs by buffering the extracellular tumour environment and intracellular acidic spaces [112]. For example, CQ can reduce the endosomal sequestration of certain drugs by raising the endosomal pH and, thus, increase their efficacy (e.g. doxorubicin, daunorubicin and mitoxantrone) [122–124]. Vezmar et al [125, 126] suggested that CQ influences multidrug resistance protein-mediated doxorubicin resistance by binding the multidrug resistance protein.
Prediction of efficacy in individual patients
Autophagy dependency and metabolic stress levels of tumour cells vary widely depending on the tumour type and progression stage. Therefore, reliable measurements to predict tumour sensitivity to autophagy inhibition would be extremely useful for patient selection in clinical practice [67]. As mentioned earlier, the status of tumour suppressor p53 can affect CQ efficacy, but other CQ sensitivity indicators have been identified as well.
First, EGFR overexpressing tumour cells, high levels of STAT3 activity, loss of caveolin-1, Akt- and Myc- driven tumour cells, and argininosuccinate synthetase enzyme deficiency are all associated with a high autophagy dependency and are therefore more sensitive to CQ administration [16, 28, 119, 127–129]. Next, there is still discussion about the effect of the oncogenic BRAF (V600E) mutation on autophagy dependency of tumour cells [47, 130, 131]. In addition, evidence has shown that autophagy is induced by the tumour suppressor alternative reading frame, but it should still be clarified whether this is cytotoxic or protective autophagy before we can determine whether CQ administration would exert beneficial effects [132]. Moreover, oncogenic Ras, and especially Kras, mutation has also been suggested as an indicator of autophagy dependency and susceptibility to CQ [27, 133], but two other studies have reported that this mutation is not a reliable indicator [134, 135]. As mentioned earlier, however, HCQ has been shown to promote tumour growth in Ras-driven pancreatic tumours developing without p53 (KrasG12D/ p53–/–) [35, 36]. Cells with the IDH1/2 mutations are metabolically vulnerable to CQ treatment, because they depend on glutaminolysis and autophagy, which is inhibited by CQ [97].
Autophagy dependency is higher in case of nutritional stress, as shown in mesothelioma cells [128], and neuroendocrine lung tumour cells are more sensitive to autophagy inhibition than non-neuroendocrine lung tumour cells [136].
Finally, an in vitro study in four human glioma cell lines observed that higher steady-state mitochondrial membrane potential values, representing mitochondrial stability, can predict cancer cell resistance to CQ treatment [137].
Our take
The final goal of this literature review was to inform further research and trials on repurposing CQ and HCQ as anti-cancer agents, as done previously for other agents [138]. In addition, the ideal dose, route of administration, and therapeutic schedule that should be applied in anti-cancer therapy was explored. Finally, the potential difference in efficacy and toxicity between CQ and HCQ has been investigated.
Efficacy of CQ and HCQ in anti-cancer therapy
The vast majority of preclinical studies on the effect of CQ monotherapy in cancer have reported a positive therapeutic effect, but the study parameters, doses, animal models and tumour types differ strongly between studies, complicating the interpretation of the results. Preclinical studies investigating the effect of HCQ in cancer are limited. Therefore, follow-up in vivo studies are warranted. A risk of publication bias exists so we cannot guarantee that all negative results have been reported.
Combination therapy with CQ or HCQ and existing anti-cancer therapies has been extensively studied in preclinical research, both in vitro and in vivo. The majority of these studies have reported an improved therapeutic efficacy as compared with monotherapy with existing anti-cancer drugs. Most studies hypothesise that CQ and HCQ could increase the efficacy of other anti-cancer drugs by blocking pro-survival autophagy. Because not all studies measured autophagy levels in vivo, it is difficult to determine to what extent the other proposed mechanisms play a role. Table 2 is limited to studies that tested CQ or HCQ in combination with conventional anti-cancer agents in vivo, but there are many other combinations that have only been tested in vitro.
Finally, multiple clinical trials have investigated, or are going to investigate, the use of CQ and HCQ in different cancer types, always in combination with other anti-cancer drugs. The availability of clinical results is limited now, as most trials are still recruiting or ongoing, and those that have been completed focused primarily on safety and tolerability of CQ and HCQ in cancer. In short, these drugs have been found safe and tolerable in all completed studies and the anti-cancer effect of both compounds is promising. However, as many clinical trials are still ongoing, a definite conclusion on the repurposing intent of CQ and HCQ in anti-cancer therapy is pending. Still, data from first clinical trials and additional preclinical data point to a potential positive implementation of these drugs in anti-cancer treatment.
Doses, route of administration and therapeutic schedule
In preclinical experiments, varying CQ and HCQ doses have been used, but most of the applied doses can be extrapolated to human doses. However, whether the dose to achieve autophagy inhibition, induction of apoptosis and tumour normalisation is achievable in humans remains an open question that would require collecting additional data in humans [112]. Clinical trials have shown that daily doses between 150 and 500 mg for CQ and daily doses between 400 and 1200 mg for HCQ are safe and well tolerated, but two studies identified 600-mg HCQ daily as the MTD. HCQ is often administered twice daily to limit plasma fluctuations and toxicity. Of note, Pascolo recommended 10 mg/kg as the maximum realistic clinical dosage of CQ, but the recommended dose and MTD of CQ and HCQ might vary dependent on the tumour type and the concomitantly administered anti-cancer treatments.
Pascolo also suggests that timing of administration is of great importance. CQ must be administered after chemotherapy and not before, which is supported by data in a mouse model of colorectal cancer treated with gemcitabine [139].
CQ or HCQ?
HCQ has been reported to have less side effects than CQ (e.g. less risk of retinal toxicity) [9, 140, 141], so it can be administered in higher doses for human use. Currently it is not clear yet whether there are differences in anti-cancer treatment efficacy between CQ and HCQ. The clinical trials that have already been completed suggest that CQ might be more efficacious than HCQ. However, no comparative clinical trial has been set up to confirm this hypothesis.
Yet, based on chemical structure, the altered safety and efficacy can be ascribed to the additional hydroxyl group in HCQ, causing pharmacokinetic differences that are essential for the working mechanism of the drugs (e.g. pKa alteration leading to differences in biprotonation and distribution) [14, 140].
Next steps
More than 30 clinical trials are currently ongoing (Feb 2017). The results of these trials may indicate which tumour types are most sensitive to CQ and HCQ treatment, and which combination therapies can be beneficial. Additional preclinical studies could further characterise the most relevant mechanisms of action and their individual importance in anti-cancer therapy. Finally, CQ analogues and other more specific autophagy inhibitory agents are also under investigation for the treatment of cancer patients (e.g. Lys05) [142–146].
Conclusion
CQ and HCQ have been studied in multiple preclinical cancer models and have demonstrated activity on several cancer-supporting pathways and in combination with a broad range of other therapies. Our review has highlighted the interesting multi-faceted actions of CQ and HCQ against cancer, making these drugs attractive for this complex disease [147, 148].
Even though it is too soon to make definite conclusions about the overall effect of CQ and HCQ in anti-cancer treatments, the clinical data already available are encouraging to further explore their potential as anti-cancer agents, with a preference for CQ. Until now, most clinical evidence was found in patients with glioblastoma and brain metastases and in patients with BRAF mutations, but some promising effects have been reported in patients with lung cancer, multiple myeloma and sarcoma as well. Although the side effects of CQ and HCQ are minor in comparison with conventional anti-cancer therapy, the possibility of retinal toxicity in trials planning long-term CQ and HCQ exposure requires the implementation of ophthalmologic monitoring. More than 30 clinical studies are currently evaluating HCQ and CQ in different cancers, most of them with the rationale to increase the efficacy of other anti-cancer therapies through inhibition of treatment-induced autophagy. The first clinical trials with CQ and HCQ have focused on the toxicity of different CQ doses in multiple populations and new trials should now focus on rigorous evaluation of efficacy.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Author contributions
Primary authors: Ciska Verbaanderd and Gauthier Bouche. Contributing authors: Hannelore Maes, Marco Schaaf, Vikas P Sukhatme, Pan Pantziarka, Vidula Sukhatme and Patrizia Agostinis. All authors read and approved the final manuscript.
References
1. Ben-Zvi I et al (2012) Hydroxychloroquine: from malaria to autoimmunity Clinical Reviews in Allergy and Immunology 42 145–153 https://doi.org/10.1007/s12016-010-8243-x
2. Thomé R et al (2013) Chloroquine: modes of action of an undervalued drug Immunology Letters 153(1–2) 50–57 https://doi.org/10.1016/j.imlet.2013.07.004 PMID: 23891850
3. Anon (n.d.) Avloclor tablets - summary of product characteristics (SPC) - (eMC) [online] Available from: https://www.medicines.org.uk/emc/medicine/2272 (Accessed 15 January 2016)
4. Anon (n.d.) Plaquenil 200mg film-coated tablets - summary of product characteristics (SPC) - (eMC) [online] Available from: https://www.medicines.org.uk/emc/medicine/6977 (Accessed 15 January 2016)
5. Lee S-J et al (2011) The role of antimalarial agents in the treatment of SLE and lupus nephritis Nature Reviews Nephrology 7(12) 718–729 https://doi.org/10.1038/nrneph.2011.150 PMID: 22009248
6. Dubois EL (1978) Antimalarials in the management of discoid and systemic lupus erythematosus Seminars in Arthritis and Rheumatism 8(1) 33–51 https://doi.org/10.1016/0049-0172(78)90033-1 PMID: 358397
7. Olson NY and Lindsley CB (1989) Adjunctive use of hydroxychloroquine in childhood dermatomyositis The Journal of Rheumatology 16(12) 1545–1547 PMID: 2483176
8. Costedoat-Chalumeau N et al (2007) Cardiomyopathy related to antimalarial therapy with illustrative case report Cardiology 107(2) 73–80 https://doi.org/10.1159/000094079
9. Browning DJ (2014) Pharmacology of chloroquine and hydroxychloroquine Hydroxychloroquine and Chloroquine Retinopathy 35–63 https://doi.org/10.1007/978-1-4939-0597-3_2
10. Costedoat-Chalumeau N et al (2015) A critical review of the effects of hydroxychloroquine and chloroquine on the eye Clinical Reviews in Allergy & Immunology 49(3) 317–326 https://doi.org/10.1007/s12016-015-8469-8
11. Nagaratnam N et al (1978) Aplasia and leukaemia following chloroquine therapy Postgraduate Medical Journal 54(628) 108–112 https://doi.org/10.1136/pgmj.54.628.108 PMID: 273209 PMCID: 2425073
12. Goyal V and Bordia A (1995) The hypoglycemic effect of chloroquine The Journal of the Association of Physicians of India 43(1) 17–18 PMID: 9282631
13. Marmor MF et al (2016) Recommendations on screening for chloroquine and hydroxychloroquine retinopathy (2016 revision) Ophthalmology 123(6) 1386–1394 https://doi.org/10.1016/j.ophtha.2016.01.058 PMID: 26992838
14. Warhurst DC et al (2003) Hydroxychloroquine is much less active than chloroquine against chloroquine-resistant plasmodium falciparum, in agreement with its physicochemical properties The Journal of Antimicrobial Chemotherapy 52(2) 188–193 https://doi.org/10.1093/jac/dkg319 PMID: 12837731
15. Day RO et al (eds.) (2005) Antirheumatic Therapy: Actions and Outcomes Birkhäuser: Basel
16. Jutten B et al (2013) EGFR overexpressing cells and tumors are dependent on autophagy for growth and survival Radiotherapy and Oncology 108(3) 479–483 https://doi.org/10.1016/j.radonc.2013.06.033 PMID: 23891088
17. Kim EL et al (2010) Chloroquine activates the p53 pathway and induces apoptosis in human glioma cells Neuro-Oncology 12(4) 389–400 https://doi.org/10.1093/neuonc/nop046 PMID: 20308316 PMCID: 2940600
18. Song YJ et al (2013) Autophagy contributes to the survival of CD133 liver cancer stem cells in the hypoxic and nutrient-deprived tumor microenvironment Cancer Letters 339(1) 70–81 https://doi.org/10.1016/j.canlet.2013.07.021 PMID: 23879969
19. Hu T et al (2016) Chloroquine inhibits hepatocellular carcinoma cell growth in vitro and in vivo Oncology Reports 35(1) 43–49 https://doi.org/10.3892/or.2015.4380 PMCID: 4699623
20. Lakhter AJ et al (2013) Chloroquine promotes apoptosis in melanoma cells by inhibiting BH3 domain-mediated PUMA degradation The Journal of Investigative Dermatology 133(9) 2247–2254 https://doi.org/10.1038/jid.2013.56 PMID: 23370537 PMCID: 3675185
21. Zheng Y et al (2009) Chloroquine inhibits colon cancer cell growth in vitro and tumor growth in vivo via induction of apoptosis Cancer Investigation 27(3) 286–292 https://doi.org/10.1080/07357900802427927 PMID: 19194831
22. Jiang PD et al (2010) Antitumor and antimetastatic activities of chloroquine diphosphate in a murine model of breast cancer Biomedicine and Pharmacotherapy 64(9) 609–614 https://doi.org/10.1016/j.biopha.2010.06.004 PMID: 20888174
23. Loehberg CR et al (2007) Ataxia telangiectasia-mutated and p53 are potential mediators of chloroquine-induced resistance to mammary carcinogenesis Cancer Research 67(24) 12026–12033 https://doi.org/10.1158/0008-5472.CAN-07-3058 PMID: 18089834
24. Maclean KH et al (2008) Targeting lysosomal degradation induces p53-dependent cell death and prevents cancer in mouse models of lymphomagenesis The Journal of Clinical Investigation 118(1) 79–88 https://doi.org/10.1172/JCI33700
25. Sun K et al (2013) Paradoxical role of autophagy in the dysplastic and tumor-forming stages of hepatocarcinoma development in rats Cell Death & Disease 4 e501 https://doi.org/10.1038/cddis.2013.35
26. Maes H et al (2014) Tumor vessel normalization by chloroquine independent of autophagy Cancer Cell 26(2) 190–206 https://doi.org/10.1016/j.ccr.2014.06.025 PMID: 25117709
27. Yang S et al (2011) Pancreatic cancers require autophagy for tumor growth Genes and Development 25(7) 717–729 https://doi.org/10.1101/gad.2016111 PMID: 21406549 PMCID: 3070934
28. Maycotte P et al (2015) STAT3-mediated autophagy dependence identifies subtypes of breast cancer where autophagy inhibition can be efficacious Cancer Research 74(9) 2579–2590 https://doi.org/10.1158/0008-5472.CAN-13-3470
29. Hiraki K and Kimura I (1963) Studies on the treatment of malignant tumors with fibroblast-inhibiting agent. II. Effects of chloroquine on animal tumors Acta Medicinae Okayama 17 239–252 PMID: 14164121
30. Pellegrini P et al (2014) Acidic extracellular pH neutralizes the autophagy-inhibiting activity of chloroquine: implications for cancer therapies Autophagy 10(4) 562–571 https://doi.org/10.4161/auto.27901 PMID: 24492472 PMCID: 3984580
31. Rouschop KMA et al (2010) The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5 The Journal of Clinical Investigation 120(1) 127–141 https://doi.org/10.1172/JCI40027 PMCID: 2798689
32. Dutta P et al (1994) Enhanced growth of mammary adenocarcinoma in rats by chloroquine and quinacrine Cancer Letters 76 113–119 https://doi.org/10.1016/0304-3835(94)90386-7 PMID: 8149339
33. Yamaguchi I et al (1966) Influence of host conditions upon the growth pattern of transplanted tumors Tohoku Journal of Experimental Medicine 90 291–301 https://doi.org/10.1620/tjem.90.291 PMID: 5971610
34. Chi C et al (2010) Disruption of lysosome function promotes tumor growth and metastasis in drosophila The Journal of Biological Chemistry 285(28) 21817–21823 https://doi.org/10.1074/jbc.M110.131714 PMID: 20418542 PMCID: 2898421
35. Rosenfeldt MT et al (2013) P53 status determines the role of autophagy in pancreatic tumour development Nature 504(7479) 296–300 https://doi.org/10.1038/nature12865 PMID: 24305049
36. Jonckheere N et al (2014) Of autophagy and in vivo pancreatic carcinogenesis: the p53 status matters! Clinics and Research in Hepatology and Gastroenterology 38(4) 423–425 https://doi.org/10.1016/j.clinre.2014.04.009 PMID: 24939064
37. Keith CT et al (2005) Multicomponent therapeutics for networked systems Nature Reviews Drug Discovery 4(1) 71–78 https://doi.org/10.1038/nrd1609 PMID: 15688074
38. Briceño E et al (2003) Therapy of glioblastoma multiforme improved by the antimutagenic chloroquine Neurosurgical Focus 14(2) e3 https://doi.org/10.3171/foc.2003.14.2.4
39. Sotelo J et al (2006) Adding chloroquine to conventional treatment for glioblastoma multiforme: a randomized, double-blind, placebo-controlled trial Annals of Internal Medicine 144 337–343 https://doi.org/10.7326/0003-4819-144-5-200603070-00008 PMID: 16520474
40. Gilbert MR (2006) New treatments for malignant gliomas: careful evaluation and cautious optimism required Annals of Iinternal Medicine 144(5) 371–373 https://doi.org/10.7326/0003-4819-144-5-200603070-00015
41. Briceño E et al (2007) Institutional experience with chloroquine as an adjuvant to the therapy for glioblastoma multiforme Surgical Neurology 67(4) 388–391 https://doi.org/10.1016/j.surneu.2006.08.080 PMID: 17350410
42. Bilger A et al (2014) FET-PET-based reirradiation and chloroquine in patients with recurrent glioblastoma: first tolerability and feasibility results Strahlentherapie und Onkologie 190(10) 957–961 https://doi.org/10.1007/s00066-014-0693-2 PMID: 24928248
43. Rojas-Puentes LL et al (2013) Phase II randomized, double-blind, placebo-controlled study of whole-brain irradiation with concomitant chloroquine for brain metastases Radiation Oncology (London, England) 8 209 https://doi.org/10.1186/1748-717X-8-209
44. Eldredge HB et al (2013) Concurrent whole brain radiotherapy and short-course chloroquine in patients with brain metastases: a pilot trial Journal of Radiation Oncology 2(3) 315–321 https://doi.org/10.1007/s13566-013-0111-x
45. Munshi A et al (2008) Unusual intensification of skin reactions by chloroquine use during breast radiotherapy Acta Oncologica (Stockholm, Sweden) 47(2) 318–319 https://doi.org/10.1080/02841860701491058
46. Rustogi A et al (2006) Unexpected skin reaction induced by radiotherapy after chloroquine use The Lancet. Oncology 7(7) 608–609 https://doi.org/10.1016/S1470-2045(06)70763-X PMID: 16814214
47. Levy JMM et al (2014) Autophagy inhibition improves chemosensitivity in BRAFV600E brain tumors Cancer Discovery 4(7) 773–780 https://doi.org/10.1158/2159-8290.CD-14-0049 PMID: 24823863 PMCID: 4090283
48. Mulcahy Levy JM et al (2017) Autophagy inhibition overcomes multiple mechanisms of resistance to BRAF inhibition in brain tumors eLife 6 358–367 https://doi.org/10.7554/eLife.19671
49. Montanari F et al (2014) A phase II trial of chloroquine in combination with bortezomib and cyclophosphamide in patients with relapsed and refractory multiple myeloma Blood 124(21) 5775
50. Kyle RA et al (1975) Multiple myeloma resistant to melphalan (NSC-8806) treated with cyclophosphamide (NSC-26271), prednisone (NSC-10023), and chloroquine (NSC-187208) Cancer Chemotherapy Reports. Part 1 59(3) 557–562 PMID: 1203882
51. Rangwala R et al (2014) Combined MTOR and autophagy inhibition: phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma Autophagy 10(8) 1391–1402 https://doi.org/10.4161/auto.29119 PMID: 24991838 PMCID: 4203516
52. Rangwala R et al (2014) Phase I trial of hydroxychloroquine with dose-intense temozolomide in patients with advanced solid tumors and melanoma Autophagy 10(8) 1369–1379 https://doi.org/10.4161/auto.29118 PMID: 24991839 PMCID: 4203514
53. Mahalingam D et al (2014) Combined autophagy and HDAC inhibition: a phase I safety, tolerability, pharmacokinetic, and pharmacodynamic analysis of hydroxychloroquine in combination with the HDAC inhibitor vorinostat in patients with advanced solid tumors Autophagy 10(8) 1403–1414 https://doi.org/10.4161/auto.29231 PMID: 24991835 PMCID: 4203517
54. Chi K-H et al (2015) Addition of rapamycin and hydroxychloroquine to metronomic chemotherapy as a second line treatment results in high salvage rates for refractory metastatic solid tumors: a pilot safety and effectiveness analysis in a small patient cohort Oncotarget 6(18) 16735–16745 https://doi.org/10.18632/oncotarget.3793 PMID: 25944689 PMCID: 4599303
55. Rosenfeld MR et al (2014) A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme Autophagy 10(8) 1359–1368 https://doi.org/10.4161/auto.28984 PMID: 24991840 PMCID: 4203513
56. Stupp R et al (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma The New England Journal of Medicine 352(10) 987–996 https://doi.org/10.1056/NEJMoa043330 PMID: 15758009
57. Goldberg SB et al (2012) A phase I study of erlotinib and hydroxychloroquine in advanced non-small-cell lung cancer Journal of Thoracic Oncology 7(10) 1602–1608 https://doi.org/10.1097/JTO.0b013e318262de4a PMID: 22878749 PMCID: 3791327
58. Leung L-SB et al (2015) Rapid onset of retinal toxicity from high-dose hydroxychloroquine given for cancer therapy American Journal of Ophthalmology 160(4) 799.e1–805.e1 https://doi.org/10.1016/j.ajo.2015.07.012
59. Vogl DT et al (2014) Combined autophagy and proteasome inhibition: a phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma Autophagy 10(8) 1380–1390 https://doi.org/10.4161/auto.29264 PMID: 24991834 PMCID: 4203515
60. Boone BA et al (2015) Safety and biologic response of pre-operative autophagy inhibition in combination with gemcitabine in patients with pancreatic adenocarcinoma Annals of Surgical Oncology 22(13) 4402–4410 https://doi.org/10.1245/s10434-015-4566-4 PMID: 25905586 PMCID: 4663459
61. Wolpin BM et al (2014) Phase II and pharmacodynamic study of autophagy inhibition using hydroxychloroquine in patients with metastatic pancreatic adenocarcinoma The Oncologist 19(6) 637–638 https://doi.org/10.1634/theoncologist.2014-0086 PMID: 24821822 PMCID: 4041680
62. Chi M-S et al (2015) Double autophagy modulators reduce 2-deoxyglucose uptake in sarcoma patients Oncotarget 6(30) 29808–29817 https://doi.org/10.18632/oncotarget.5060 PMID: 26375670 PMCID: 4745764
63. Glick D et al (2010) Autophagy: cellular and molecular mechanisms The Journal of Pathology 221(1) 3–12 https://doi.org/10.1002/path.2697 PMID: 20225336 PMCID: 2990190
64. Sehgal A et al (2014) You eat what you are: autophagy inhibition as a therapeutic strategy in leukemia Leukemia 29(3) 517–525 https://doi.org/10.1038/leu.2014.349 PMID: 25541151 PMCID: 4825874
65. Parzych KR and Klionsky DJ (2014) An overview of autophagy: morphology, mechanism, and regulation Antioxidants & Redox Signaling 20(3) 460–473 https://doi.org/10.1089/ars.2013.5371
66. Viry E et al (2014) Autophagy: an adaptive metabolic response to stress shaping the antitumor immunity Biochemical Pharmacology 92(1) 1–12 https://doi.org/10.1016/j.bcp.2014.07.006
67. Cicchini M et al (2015) Molecular pathways: autophagy in cancer–a matter of timing and context Clinical Cancer Research 21(3) 498–504 https://doi.org/10.1158/1078-0432.CCR-13-2438
68. Cheong H (2015) Integrating autophagy and metabolism in cancer Archives of Pharmacal Research 38(3) 358–371 https://doi.org/10.1007/s12272-015-0562-2 PMID: 25614051
69. Janku F et al (2011) Autophagy as a target for anticancer therapy Nature Reviews Clinical Oncology 8(9) 528–539 https://doi.org/10.1038/nrclinonc.2011.71 PMID: 21587219
70. Rebecca VW and Amaravadi RK (2016) Emerging strategies to effectively target autophagy in cancer Oncogene 35(1) 1–11 https://doi.org/10.1038/onc.2015.99 PMCID: 4838040
71. Townsend KN et al (2012) Autophagy inhibition in cancer therapy: metabolic considerations for antitumor immunity Immunological Reviews 249(1) 176–194 https://doi.org/10.1111/j.1600-065X.2012.01141.x PMID: 22889222
72. Loehberg CR et al (2012) Akt and p53 are potential mediators of reduced mammary tumor growth by cloroquine and the mTOR inhibitor RAD001 Biochemical Pharmacology 83(4) 480–488 https://doi.org/10.1016/j.bcp.2011.11.022
73. Zhang Y et al (2014) Functional expression of TLR9 in esophageal cancer Oncology Reports 31(5) 2298–2304. https://doi.org/10.3892/or.2014.3095 PMID: 24647486
74. Zhang Y et al (2015) Chloroquine inhibits MGC803 gastric cancer cell migration via the Toll-like receptor 9/nuclear factor kappa B signaling pathway Molecular Medicine Reports 11(2) 1366–1371 https://doi.org/10.3892/mmr.2014.2839
75. Väisänen MR et al (2013) Expression of toll-like receptor-9 is associated with poor progression-free survival in prostate cancer Oncology Letters 5(12) 1659–1663 PMID: 23761830 PMCID: 3678868
76. Mohamed FE et al (2015) Effect of toll-like receptor 7 and 9 targeted therapy to prevent the development of hepatocellular carcinoma Liver International 35(3) 1063–1076 https://doi.org/10.1111/liv.12626
77. Kuznik A et al (2011) Mechanism of endosomal TLR inhibition by antimalarial drugs and imidazoquinolines The Journal of Immunology 186(8) 4794–4804 https://doi.org/10.4049/jimmunol.1000702 PMID: 21398612
78. Sandholm J et al (2014) Hypoxia regulates toll-like receptor-9 expression and invasive function in human brain cancer cells in vitro Oncology Letters 8 266–274 PMID: 24959259 PMCID: 4063648
79. Tuomela J et al (2012) Low TLR9 expression defines an aggressive subtype of triple-negative breast cancer Breast Cancer Research and Treatment 135 481–493 https://doi.org/10.1007/s10549-012-2181-7 PMID: 22847512
80. Tuomela J et al (2013) Chloroquine has tumor-inhibitory and tumor-promoting effects in triple-negative breast cancer Oncology Letters 6 1665–1672 PMID: 24273604 PMCID: 3835157
81. Sun X et al (2010) CXCL12 / CXCR4 / CXCR7 chemokine axis and cancer progression Cancer Metastasis Reviews 29(4) 709–722 https://doi.org/10.1007/s10555-010-9256-x PMID: 20839032 PMCID: 3175097
82. Kim J et al (2012) Identification of anti-malarial compounds as novel antagonists to chemokine receptor CXCR4 in pancreatic cancer cells PloS One 7(2) e31004 https://doi.org/10.1371/journal.pone.0031004 PMID: 22319600 PMCID: 3272047
83. Balic A et al (2014) Chloroquine targets pancreatic cancer stem cells via inhibition of CXCR4 and hedgehog signaling Molecular Cancer Therapeutics 13(7) 1758–1771 https://doi.org/10.1158/1535-7163.MCT-13-0948 PMID: 24785258
84. Bieging KT et al (2014) Unravelling mechanisms of p53-mediated tumour suppression Nature Reviews Cancer 14(5) 359–370 https://doi.org/10.1038/nrc3711 PMID: 24739573 PMCID: 4049238
85. Zhou Q et al (2002) Control of mammary tumor cell growth in vitro by novel cell differentiation and apoptosis agents Breast Cancer Research and Treatment 75(2) 107–117 https://doi.org/10.1023/A:1019698807564 PMID: 12243503
86. Lee SW et al (2015) The synergistic effect of combination temozolomide and chloroquine treatment is dependent on autophagy formation and p53 status in glioma cells Cancer Letters 360(2) 195–204 https://doi.org/10.1016/j.canlet.2015.02.012 PMID: 25681668
87. Amaravadi R and Debnath J (2014) Mouse models address key concerns regarding autophagy inhibition in cancer therapy Cancer Discovery 4(8) 873–875 https://doi.org/10.1158/2159-8290.CD-14-0618 PMID: 25092744 PMCID: 4124512
88. Yang A and Kimmelman AC (2014) Inhibition of autophagy attenuates pancreatic cancer growth independent of TP53/TRP53 status Autophagy 10(9) 1683–1684 https://doi.org/10.4161/auto.29961 PMID: 25046107 PMCID: 4206544
89. Liu F et al (2014) Chloroquine potentiates the anti-cancer effect of lidamycin on non-small cell lung cancer cells in vitro Acta Pharmacologica Sinica 35(5) 645–652 https://doi.org/10.1038/aps.2014.3 PMID: 24727941 PMCID: 4814038
90. Geng Y et al (2010) Chloroquine-induced autophagic vacuole accumulation and cell death in glioma cells is p53 independent Neuro-Oncology 12(5) 473–481 PMID: 20406898 PMCID: 2940627
91. Burikhanov R et al (2017) Chloroquine-inducible par-4 secretion is essential for tumor cell apoptosis and inhibition of metastasis Cell Reports 18(2) 508–519 https://doi.org/10.1016/j.celrep.2016.12.051 PMID: 28076793 PMCID: 5264245
92. Amaravadi RK (2013) PUMA: a puzzle piece in chloroquine’s antimelanoma activity The Journal of Investigative Dermatology 133(9) 2133–2135 https://doi.org/10.1038/jid.2013.135 PMID: 23949767 PMCID: 4825873
93. Jin L et al (2016) Glutaminolysis as a target for cancer therapy Oncogene 35(28) 3619–3625 https://doi.org/10.1038/onc.2015.447
94. Choi M-M et al (2007) Inhibitory properties of nerve-specific human glutamate dehydrogenase isozyme by chloroquine Journal of Biochemistry and Molecular Biology 40(6) 1077–1082 PMID: 18047806
95. Jarzyna R et al (1997) Chloroquine is a potent inhibitor of glutamate dehydrogenase in liver and kidney-cortex of rabbit Pharmacological Research : The Official Journal of the Italian Pharmacological Society 35(1) 79–84 https://doi.org/10.1006/phrs.1996.0108
96. Jarzyna R et al (2001) The inhibition of gluconeogenesis by chloroquine contributes to its hypoglycaemic action European Journal of Pharmacology 428(3) 381–388 https://doi.org/10.1016/S0014-2999(01)01221-3 PMID: 11689198
97. Molenaar RJ et al (2017) Study protocol of a phase IB/II clinical trial of metformin and chloroquine in patients with IDH1-mutated or IDH2-mutated solid tumours BMJ Open 7(6) e014961 https://doi.org/10.1136/bmjopen-2016-014961 PMID: 28601826 PMCID: 5541450
98. Lagneaux L et al (2001) Early induction of apoptosis in B-chronic lymphocytic leukaemia cells by hydroxychloroquine: activation of caspase-3 and no protection by survival factors British Journal of Haematology 112(2) 344–352 https://doi.org/10.1046/j.1365-2141.2001.02553.x PMID: 11167827
99. Lagneaux L et al (2002) Hydroxychloroquine-induced apoptosis of chronic lymphocytic leukemia involves activation of caspase-3 and modulation of Bcl-2/bax/ratio Leukemia & Lymphoma 43(5) 1087–1095 https://doi.org/10.1080/10428190290021506
100. Park BC et al (2008) Chloroquine-induced nitric oxide increase and cell death is dependent on cellular GSH depletion in A172 human glioblastoma cells Toxicology Letters 178(1) 52–60 https://doi.org/10.1016/j.toxlet.2008.02.003 PMID: 18359172
101. Jiang P et al (2008) Cell growth inhibition, G2/M cell cycle arrest, and apoptosis induced by chloroquine in human breast cancer cell line Bcap-37 Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology 22(5–6) 431–440 https://doi.org/10.1159/000185488 PMID: 19088425
102. Fan C et al (2006) Chloroquine inhibits cell growth and induces cell death in A549 lung cancer cells Bioorganic & Medicinal Chemistry 14(9) 3218–3222 https://doi.org/10.1016/j.bmc.2005.12.035
103. Rahim R and Strobl JS (2009) Hydroxychloroquine, chloroquine, and all-trans retinoic acid regulate growth, survival, and histone acetylation in breast cancer cells Anti-Cancer Drugs 20(8) 736–745 https://doi.org/10.1097/CAD.0b013e32832f4e50 PMID: 19584707
104. Justilien V and Fields AP (2015) Molecular pathways: novel approaches for improved therapeutic targeting of hedgehog signaling in cancer stem cells Clinical Cancer Research 21 505–513 https://doi.org/10.1158/1078-0432.CCR-14-0507 PMID: 25646180 PMCID: 4316382
105. Thongchot S et al (2015) Chloroquine exerts anti-metastatic activities under hypoxic conditions in cholangiocarcinoma cells Asian Pacific Journal of Cancer Prevention : APJCP 16(5) 2031–2035 https://doi.org/10.7314/APJCP.2015.16.5.2031 PMID: 25773848
106. Choi DS et al (2014) Chloroquine eliminates cancer stem cells through deregulation of Jak2 and DNMT1 Stem Cells (Dayton, Ohio) 32(9) 2309–2323 https://doi.org/10.1002/stem.1746
107. Liang DH et al (2016) The autophagy inhibitor chloroquine targets cancer stem cells in triple negative breast cancer by inducing mitochondrial damage and impairing DNA break repair Cancer Letters 376(2) 249–258 https://doi.org/10.1016/j.canlet.2016.04.002 PMID: 27060208 PMCID: 4864217
108. Inoue S et al (1993) Antimelanoma activity of chloroquine, an antimalarial agent with high affinity for melanin Pigment Cell Research / Sponsored by the European Society for Pigment Cell Research and the International Pigment Cell Society, 6(5), pp. 354–8. https://doi.org/10.1111/j.1600-0749.1993.tb00613.x PMID: 8302774
109. Karmali RA et al (1978) Chloroquine enhances Epstein–Barr virus expression Nature 275(5679) 444–445 https://doi.org/10.1038/275444a0 PMID: 211434
110. Lenoir G and Geser A (1979) Effect of chloroquine on Epstein–Barr virus expression Nature 282(5740) 758 https://doi.org/10.1038/282758b0 PMID: 229421
111. Mellman I et al (2011) Cancer immunotherapy comes of age Nature 480(7378) 480–489 https://doi.org/10.1038/nature10673 PMID: 22193102 PMCID: 3967235
112. Pascolo S (2016) Time to use a dose of chloroquine as an adjuvant to anti-cancer chemotherapies European Journal of Pharmacology 771 139–144 https://doi.org/10.1016/j.ejphar.2015.12.017
113. Bergers G and Benjamin LE (2003) Angiogenesis: tumorigenesis and the angiogenic switch Nature Reviews Cancer 3(6) 401–410 https://doi.org/10.1038/nrc1093 PMID: 12778130
114. Jain RK (2005) Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy Science (New York, NY) 307(5706) 58–62 https://doi.org/10.1126/science.1104819
115. Carmeliet P and Jain RK (2011) Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases Nature Reviews Drug Discovery 10(6) 417–427 https://doi.org/10.1038/nrd3455 PMID: 21629292
116. Maes H et al (2014) How to teach an old dog new tricks: autophagy-independent action of chloroquine on the tumor vasculature Autophagy 10(11) 2082–2084 https://doi.org/10.4161/auto.36259 PMID: 25484095 PMCID: 4502691
117. Hagihara N et al (2000) Vascular protection by chloroquine during brain tumor therapy with Tf-CRM107 advances in brief vascular protection by chloroquine during brain tumor therapy with Tf-CRM107 Cancer Research 60(2) 230–234 PMID: 10667564
118. McAllister SS and Weinberg RA (2014) The tumour-induced systemic environment as a critical regulator of cancer progression and metastasis Nature Cell Biology 16(8) 717–727 https://doi.org/10.1038/ncb3015 PMID: 25082194
119. Ko Y-H et al (2011) Glutamine fuels a vicious cycle of autophagy in the tumor stroma and oxidative mitochondrial metabolism in epithelial cancer cells: implications for preventing chemotherapy resistance Cancer Biology & Therapy 12(12) 1085–1097 https://doi.org/10.4161/cbt.12.12.18671
120. Martinez-Outschoorn UE et al (2010) Tumor cells induce the cancer associated fibroblast phenotype via caveolin-1 degradation: implications for breast cancer and DCIS therapy with autophagy inhibitors Cell Cycle (Georgetown, TX) 9(12) 2423–2433 https://doi.org/10.4161/cc.9.12.12048
121. Kimura T et al (2013) Chloroquine in cancer therapy: a double-edged sword of autophagy Cancer Research 73(1) 3–7 https://doi.org/10.1158/0008-5472.CAN-12-2464 PMID: 23288916
122. Lee CM and Tannock IF (2006) Inhibition of endosomal sequestration of basic anticancer drugs: influence on cytotoxicity and tissue penetration British Journal of Cancer 94(6) 863–869 https://doi.org/10.1038/sj.bjc.6603010 PMID: 16495919 PMCID: 2361369
123. Hurwitz SJ et al (1997) Vesicular anthracycline accumulation in doxorubicin-selected U-937 cells: participation of lysosomes Blood 89(10) 3745–3754 PMID: 9160680
124. Maycotte P et al (2012) Chloroquine sensitizes breast cancer cells to chemotherapy independent of autophagy Autophagy 8(2) 200–212 https://doi.org/10.4161/auto.8.2.18554 PMID: 22252008 PMCID: 3336076
125. Vezmar M and Georges E (1998) Direct binding of chloroquine to the multidrug resistance protein (MRP): possible role for MRP in chloroquine drug transport and resistance in tumor cells Biochemical Pharmacology 56(6) 733–742 https://doi.org/10.1016/S0006-2952(98)00217-2 PMID: 9751078
126. Vezmar M and Georges E (2000) Reversal of MRP-mediated doxorubicin resistance with quinoline-based drugs Biochemical Pharmacology 59(10) 1245–1252 https://doi.org/10.1016/S0006-2952(00)00270-7 PMID: 10736425
127. Maes H et al (2014) Dynamic interplay between autophagic flux and Akt during melanoma progression in vitro Experimental Dermatology 23(2) 101–106 https://doi.org/10.1111/exd.12298
128. Battisti S et al (2012) Nutritional stress and arginine auxotrophy confer high sensitivity to chloroquine toxicity in mesothelioma cells American Journal of Respiratory Cell and Molecular Biology 46(4) 498–506 https://doi.org/10.1165/rcmb.2011-0195OC
129. Amaravadi RK et al (2007) Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma The Journal of Clinical Investigation 117(2) 326–336 https://doi.org/10.1172/JCI28833 PMID: 17235397 PMCID: 1765515
130. Levy JM et al (2014) Using BRAF(V600E) as a marker of autophagy dependence in pediatric brain tumors Autophagy 10(11) 2077–2078 https://doi.org/10.4161/auto.36138
131. Egger ME et al (2013) Inhibition of autophagy with chloroquine is effective in melanoma The Journal of Surgical Research 184(1) 274–281 https://doi.org/10.1016/j.jss.2013.04.055 PMID: 23706562
132. Pimkina J and Murphy ME (2009) ARF, autophagy and tumor suppression Autophagy 5(3) 397–399 https://doi.org/10.4161/auto.5.3.7782 PMID: 19221462 PMCID: 2667648
133. Mancias JD and Kimmelman AC (2011) Targeting autophagy addiction in cancer Oncotarget 2(12) 1302–1306 https://doi.org/10.18632/oncotarget.384 PMID: 22185891 PMCID: 3282086
134. Morgan MJ et al (2014) Regulation of autophagy and chloroquine sensitivity by oncogenic RAS in vitro is context-dependent Autophagy 10(10) 1814–1826 https://doi.org/10.4161/auto.32135 PMID: 25136801 PMCID: 4198365
135. Eng CH et al (2016) Macroautophagy is dispensable for growth of KRAS mutant tumors and chloroquine efficacy Proceedings of the National Academy of Sciences of the United States of America 113(1) 182–187 https://doi.org/10.1073/pnas.1515617113 PMCID: 4711870
136. Hong S-K et al (2013) Autophagy sensitivity of neuroendocrine lung tumor cells International Journal of Oncology 43(6) 2031–2038 https://doi.org/10.3892/ijo.2013.2136 PMID: 24126619 PMCID: 3834067
137. Vessoni AT et al (2016) Chloroquine-induced glioma cells death is associated with mitochondrial membrane potential loss, but not oxidative stress Free Radical Biology and Medicine 90 91–100 https://doi.org/10.1016/j.freeradbiomed.2015.11.008
138. Pantziarka P et al (2014) The Repurposing Drugs in Oncology (ReDO) project Ecancer Medical Science 8 442 PMID: 25075216 PMCID: 4096030
139. Buch I et al (2016) Schedule-dependent synergy of chloroquine with chemotherapy for anti-cancer treatment Cancer Research and Oncology: Open Access 2(2)
140. Schroeder RL and Gerber JP (2014) Chloroquine and hydroxychloroquine binding to melanin: some possible consequences for pathologies Toxicology Reports 1 963–968 https://doi.org/10.1016/j.toxrep.2014.10.019 PMID: 28962308 PMCID: 5598414
141. Sundelin SP and Terman A (2002) Different effects of chloroquine and hydroxychloroquine on lysosomal function in cultured retinal pigment epithelial cells APMIS : acta pathologica, microbiologica, et immunologica Scandinavica 110(6) 481–489 https://doi.org/10.1034/j.1600-0463.2002.100606.x PMID: 12193209
142. McAfee Q et al (2012) Autophagy inhibitor Lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency Proceedings of the National Academy of Sciences of the United States of America 109(21) 8253–8258 https://doi.org/10.1073/pnas.1118193109 PMID: 22566612 PMCID: 3361415
143. Solomon VR and Lee H (2009) Chloroquine and its analogs: a new promise of an old drug for effective and safe cancer therapies European Journal of Pharmacology 625(1–3) 220–233 https://doi.org/10.1016/j.ejphar.2009.06.063 PMID: 19836374
144. Zhang H et al (2008) Synthesis and in vitro cytotoxicity evaluation of 4-aminoquinoline derivatives Biomedicine & Pharmacotherapy 62(2) 65–69 https://doi.org/10.1016/j.biopha.2007.04.007
145. Goodall ML et al (2014) Development of potent autophagy inhibitors that sensitize oncogenic BRAF V600E mutant melanoma tumor cells to vemurafenib Autophagy 10(6) 1120–1136 https://doi.org/10.4161/auto.28594 PMID: 24879157 PMCID: 4091172
146. Dai J-P et al (2013) Drug screening for autophagy inhibitors based on the dissociation of Beclin1-Bcl2 complex using BiFC technique and mechanism of eugenol on anti-influenza A virus activity PloS One 8(4) e61026 https://doi.org/10.1371/journal.pone.0061026 PMID: 23613775 PMCID: 3628889
147. Anighoro A et al (2014) Polypharmacology: challenges and opportunities in drug discovery Journal of Medicinal Chemistry 57(19) 7874–7887 https://doi.org/10.1021/jm5006463 PMID: 24946140
148. Hopkins AL (2008) Network pharmacology: the next paradigm in drug discovery Nature Chemical Biology 4(11) 682–690 https://doi.org/10.1038/nchembio.118 PMID: 18936753
149. Reagan-Shaw S et al (2008) Dose translation from animal to human studies revisited FASEB Journal : Official Publication of the Federation of American Societies for Experimental Biology 22(3) 659–661 https://doi.org/10.1096/fj.07-9574LSF
150. Golden EB et al (2014) Chloroquine enhances temozolomide cytotoxicity in malignant gliomas by blocking autophagy Neurosurgical Focus 37(6) E12 https://doi.org/10.3171/2014.9.FOCUS14504 PMID: 25434381
151. Zanotto-Filho A et al (2015) Autophagy inhibition improves the efficacy of curcumin/temozolomide combination therapy in glioblastomas Cancer Letters 358(2) 220–231 https://doi.org/10.1016/j.canlet.2014.12.044
152. Gaudin D et al (1971) The effect of DNA repair inhibitors on e response of tumors treated with x-ray and alkylating agents Proceedings of the Society for Experimental Biology and Medicine (New York, NY) 137(1) 202–206 https://doi.org/10.3181/00379727-137-35544
153. Lefort S et al (2014) Inhibition of autophagy as a new means of improving chemotherapy efficiency in high-LC3B triple-negative breast cancers Autophagy 10(12) 2122–2142 https://doi.org/10.4161/15548627.2014.981788 PMID: 25427136 PMCID: 4502743
154. Yu L et al (2014) Induction of autophagy counteracts the anticancer effect of cisplatin in human esophageal cancer cells with acquired drug resistance Cancer Letters 355 34–45 https://doi.org/10.1016/j.canlet.2014.09.020 PMID: 25236911
155. Zhang H et al (2015) Antitumor activity of chloroquine in combination with cisplatin in human gastric cancer xenografts Asian Pacific Journal of Cancer Prevention 16 3907–3912 https://doi.org/10.7314/APJCP.2015.16.9.3907 PMID: 25987058
156. Zhao X et al (2015) Chloroquine-enhanced efficacy of cisplatin in the treatment of hypopharyngeal carcinoma in xenograft mice PloS One 10(4): e01 1–12
157. Ding Z-B et al (2011) Autophagy activation in hepatocellular carcinoma contributes to the tolerance of oxaliplatin via reactive oxygen species modulation Clinical Cancer Research : An Official Journal of the American Association for Cancer Research 17(19) 6229–6238 https://doi.org/10.1158/1078-0432.CCR-11-0816 PMID: 21825039
158. Selvakumaran M et al (2013) Autophagy inhibition sensitizes colon cancer cells to antiangiogenic and cytotoxic therapy Clinical Cancer Research 19(11) 2995–3007 https://doi.org/10.1158/1078-0432.CCR-12-1542 PMID: 23461901
159. Shoemaker JP (1978) Fifty-five percent complete remission of mammary carcinoma in mice with 5-fluorouracil and chloroquine Cancer Research 38(September) 2700–2702 PMID: 679173
160. Guo X-L et al (2012) Targeting autophagy potentiates chemotherapy-induced apoptosis and proliferation inhibition in hepatocarcinoma cells Cancer Letters 320(2) 171–179 https://doi.org/10.1016/j.canlet.2012.03.002 PMID: 22406827
161. Sasaki K et al (2012) Resistance of colon cancer to 5-fluorouracil may be overcome by combination with chloroquine, an in vivo study Anti-Cancer Drugs 23(7) 675–682 https://doi.org/10.1097/CAD.0b013e328353f8c7 PMID: 22561420
162. Shoemaker JP and Dagher RK (1979) Remissions of mammary adenocarcinoma in hypothyroid mice given 5-fluorouracil and chloroquine phosphate Journal of the National Cancer Institute 62(6) 1575–1578 PMID: 286128
163. Xiong S et al (2010) Triggering liposomal drug release with a lysosomotropic agent Journal of Pharmaceutical Sciences 99(12) 5011–5018 https://doi.org/10.1002/jps.22210 PMID: 20821395
164. Arnold AM and Whitehouse JM (1982) Interaction of VP16-213 with the DNA repair antagonist chloroquine Cancer Chemotherapy and Pharmacology 7(2–3) 123–126 https://doi.org/10.1007/BF00254533
165. Cook KL et al (2014) Hydroxychloroquine inhibits autophagy to potentiate antiestrogen responsiveness in ER breast cancer Clinical Cancer Research : an Official Journal of the American Association for Cancer Research 20(12) 3222–3232 https://doi.org/10.1158/1078-0432.CCR-13-3227 PMID: 24928945 PMCID: 4073207
166. Seront E et al (2013) Tumour hypoxia determines the potential of combining mTOR and autophagy inhibitors to treat mammary tumours British Journal of Cancer 109(10) 2597–2606 https://doi.org/10.1038/bjc.2013.644 PMID: 24157830 PMCID: 3833227
167. Bray K et al (2012) Autophagy suppresses RIP kinase-dependent necrosis enabling survival to mTOR inhibition PloS One 7(7) e41831 https://doi.org/10.1371/journal.pone.0041831 PMID: 22848625 PMCID: 3406086
168. Kaneko M et al (2014) Temsirolimus and chloroquine cooperatively exhibit a potent antitumor effect against colorectal cancer cells Journal of Cancer Research and Clinical Oncology 140(5) 769–781 https://doi.org/10.1007/s00432-014-1628-0 PMID: 24619662
169. Xie X et al (2013) Coordinate autophagy and mTOR pathway inhibition enhances cell death in melanoma PloS One 8(1) e55096 https://doi.org/10.1371/journal.pone.0055096 PMID: 23383069 PMCID: 3559441
170. Rao R et al (2012) Combination of pan-histone deacetylase inhibitor and autophagy inhibitor exerts superior efficacy against triple-negative human breast cancer cells Molecular Cancer Therapeutics 11(4) 973–983 https://doi.org/10.1158/1535-7163.MCT-11-0979 PMID: 22367781
171. Carew JS et al (2010) Autophagy inhibition enhances vorinostat-induced apoptosis via ubiquitinated protein accumulation Journal of Cellular and Molecular Medicine 14(10) 2448–2459 https://doi.org/10.1111/j.1582-4934.2009.00832.x PMCID: 2891399
172. Ding W-X et al (2009) Oncogenic transformation confers a selective susceptibility to the combined suppression of the proteasome and autophagy Molecular Cancer Therapeutics 8(7) 2036–2045 https://doi.org/10.1158/1535-7163.MCT-08-1169 PMID: 19584239 PMCID: 2711219
173. Hui B et al (2012) Proteasome inhibitor interacts synergistically with autophagy inhibitor to suppress proliferation and induce apoptosis in hepatocellular carcinoma Cancer 118(22) 5560–5571 https://doi.org/10.1002/cncr.27586 PMID: 22517429
174. Tang M-C et al (2015) Chloroquine enhances gefitinib cytotoxicity in gefitinib-resistant nonsmall cell lung cancer cells PloS One 10(3) e0119135 https://doi.org/10.1371/journal.pone.0119135 PMID: 25807554 PMCID: 4373825
175. Dragowska WH et al (2013) Induction of autophagy is an early response to gefitinib and a potential therapeutic target in breast cancer PLoS One 8(10) 1–20 https://doi.org/10.1371/journal.pone.0076503
176. Bokobza SM et al (2014) Combining AKT inhibition with chloroquine and gefitinib prevents compensatory autophagy and induces cell death in EGFR mutated NSCLC cells Oncotarget 5(13) 4765–4778 https://doi.org/10.18632/oncotarget.2017 PMID: 24946858 PMCID: 4148097
177. Zou Y et al (2013) The autophagy inhibitor chloroquine overcomes the innate resistance of wild-type EGFR non-small-cell lung cancer cells to erlotinib Journal of Thoracic Oncology : Official Publication of the International Association for the Study of Lung Cancer 8(6) 693–702 https://doi.org/10.1097/JTO.0b013e31828c7210 PMID: 23575415 PMCID: 3855301
178. Bellodi C et al. (2009) Targeting autophagy potentiates tyrosine kinase inhibitor-induced cell death in Philadelphia chromosome-positive cells, including primary CML stem cells The Journal of Clinical Investigation 119(5) 1109–1123 https://doi.org/10.1172/JCI35660 PMID: 19363292 PMCID: 2673867
179. Abdel-Aziz AK et al (2014) Chloroquine synergizes sunitinib cytotoxicity via modulating autophagic, apoptotic and angiogenic machineries Chemico-Biological Interactions 217 28–40 https://doi.org/10.1016/j.cbi.2014.04.007 PMID: 24751611
180. Shimizu S et al (2012) Inhibition of autophagy potentiates the antitumor effect of the multikinase inhibitor sorafenib in hepatocellular carcinoma International Journal of Cancer 131(3) 548–557 https://doi.org/10.1002/ijc.26374
181. Shi Y-H et al (2011) Targeting autophagy enhances sorafenib lethality for hepatocellular carcinoma via ER stress-related apoptosis Autophagy 7(10) 1159–1172 https://doi.org/10.4161/auto.7.10.16818 PMID: 21691147
182. Ji C et al (2014) Induction of autophagy contributes to crizotinib resistance in ALK-positive lung cancer Cancer Biology & Therapy 15(5) 570–577 https://doi.org/10.4161/cbt.28162
183. You L et al (2015) Crizotinib induces autophagy through inhibition of the STAT3 pathway in multiple lung cancer cell lines Oncotarget 6(37) 40268–40282 https://doi.org/10.18632/oncotarget.5592 PMID: 26384345 PMCID: 4741894
184. Mitou G et al (2015) Targeting autophagy enhances the anti-tumoral action of crizotinib in ALK-positive anaplastic large cell lymphoma Oncotarget 6(30) 30149–30164 https://doi.org/10.18632/oncotarget.4999 PMID: 26338968 PMCID: 4745787
185. Shen J et al (2013) Autophagy inhibition induces enhanced proapoptotic effects of ZD6474 in glioblastoma British Journal of Cancer 109(1) 164–171 https://doi.org/10.1038/bjc.2013.306 PMID: 23799852 PMCID: 3708568
186. Hu Y-L et al (2012) Hypoxia-induced autophagy promotes tumor cell survival and adaptation to antiangiogenic treatment in glioblastoma Cancer Research 72(7) 1773–1783 https://doi.org/10.1158/0008-5472.CAN-11-3831 PMID: 22447568 PMCID: 3319869
187. Cufí S et al (2013) The anti-malarial chloroquine overcomes primary resistance and restores sensitivity to trastuzumab in HER2-positive breast cancer Scientific Reports 3 2469 https://doi.org/10.1038/srep02469 PMCID: 3749547
188. Ratikan JA et al (2013) Chloroquine engages the immune system to eradicate irradiated breast tumors in mice International Journal of Radiation Oncology, Biology, Physics 87(4) 761–768 https://doi.org/10.1016/j.ijrobp.2013.07.024 PMID: 24138918
189. Wei M-F et al (2014) Autophagy promotes resistance to photodynamic therapy-induced apoptosis selectively in colorectal cancer stem-like cells Autophagy 10(7) 1179–1192 https://doi.org/10.4161/auto.28679 PMID: 24905352 PMCID: 4203546
190. Liang X et al (2012) Inhibiting systemic autophagy during interleukin 2 immunotherapy promotes long-term tumor regression Cancer Research 72(11) 2791–2801 https://doi.org/10.1158/0008-5472.CAN-12-0320 PMID: 22472122 PMCID: 3417121
191. Thomas S et al (2012) Preferential killing of triple-negative breast cancer cells in vitro and in vivo when pharmacological aggravators of endoplasmic reticulum stress are combined with autophagy inhibitors Cancer Letters 325(1) 63–71 https://doi.org/10.1016/j.canlet.2012.05.030 PMID: 22664238
192. Harhaji-Trajkovic L et al (2012) Chloroquine-mediated lysosomal dysfunction enhances the anticancer effect of nutrient deprivation Pharmaceutical Research 29(8) 2249–2263 https://doi.org/10.1007/s11095-012-0753-1 PMID: 22538436
193. Thomas R et al (1990) The effect of chloroquine and hyperthermia on murine neuroblastoma Journal of Pediatric Surgery 25(9) 929–932 https://doi.org/10.1016/0022-3468(90)90232-X PMID: 2213444
194. Gao L et al (2013) Chloroquine promotes the anticancer effect of TACE in a rabbit VX2 liver tumor model International Journal of Biological Sciences 9(4) 322–330 https://doi.org/10.7150/ijbs.5925 PMID: 23569437 PMCID: 3619094