Tumour progression and metastasis
Francisco Arvelo1, 2, Felipe Sojo1, 2 and Carlos Cotte2
1Centro de Biociencias, Fundación Instituto de Estudios Avanzado [IDEA], Caracas 1015-A, Venezuela, Apartado 17606, Caracas 1015-A, Venezuela
2Laboratorio de Cultivo de Tejidos y Biología de Tumores, Instituto de Biología Experimental, Universidad Central de Venezuela, Apartado 47114, Caracas, 1041-A, Venezuela
Correspondence to: Francisco Arvelo. Email: franarvelo@yahoo.com
Abstract
The two biological mechanisms that determine types of malignancy are infiltration and metastasis, for which tumour microenvironment plays a key role in developing and establishing the morphology, growth and invasiveness of a malignancy. The microenvironment is formed by complex tissue containing the extracellular matrix, tumour and non-tumour cells, a signalling network of cytokines, chemokines, growth factors, and proteases that control autocrine and paracrine communication among individual cells, facilitating tumour progression. During the development of the primary tumour, the tumour stroma and continuous genetic changes within the cells makes it possible for them to migrate, having to count on a pre-metastatic niche receptor that allows the tumour’s survival and distant growth. These niches are induced by factors produced by the primary tumour; if it is eradicated, the active niches become responsible for activating the latent disseminated cells. Due to the importance of these mechanisms, the strategies that develop tumour cells during tumour progression and the way in which the microenvironment influences the formation of metastasis are reviewed. It also suggests that the metastatic niche can be an ideal target for new treatments that make controlling metastasis possible.
Keywords: cancer, infiltration, metastasis, microenvironment, metastatic niche, latency, epithelial–mesenchymal transition
Copyright: © the authors; licensee ecancermedicalscience. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Published: 29/01/2016; Received: 16/06/2015
Introduction
Metastasis is defined as ‘the process of dissemination of cancer cells from their origin to a distant organ’ [1], a complex process involving several stages, which are as follows: (a) the activation of epithelial–mesenchymal transition (EMT), during which cancer cells lose all cell–cell contact, such as substrate adhesion, acquiring ownership of movement; (b) local invasion, whereby malignant cells degrade the basal lamina, the special extracellular matrix that organises and separates epithelial tissues from the stroma, which plays an important role in both cell signalling and being a reservoir of growth factors released by tumour cells; (c) intravasation, during which tumour cells pass through the walls of blood vessels and enter the bloodstream; (d) the ability to survive in the bloodstream; (e) extravasation, whereby tumour cells exit the bloodstream, passing through the walls of blood vessels into the tissue of a particular organ; (f) establishment of tumour cells in the tissues of the organ where metastasis will form; in other words, the establishment of a pre-metastatic niche to create a favourable environment for the growth of cancer cells (Figure 1).
Each of the steps necessary in producing metastasis, from the arrival of malignant cells to their growth and proliferation in the host organ, is led by the genetic and/or epigenetic alterations acquired and accumulated during the course of tumour progression [2, 3]. Despite recent advances made in surgical techniques, radiotherapy and the development of molecularly targeted therapies, the majority of cancer-related deaths (more than 90%) are the result of progressive growth of therapy-resistant metastasis [4]. Metastatic cells come from a heterogeneous cell population within the primary tumour which, over time, are selected, experience a high spontaneous mutation rate, are more prone to suffering from rapid phenotypic diversification and are resistant to therapeutic treatments [5].
The survival of the malignant cells that form micrometastasis in the receptor organ is not ensured, as there may be differences between the primary tumour’s microenvironment and the location where the cancer cells will be established [6]. Therefore, the pre-metastatic niche model has been proposed, which can be described as ‘the location with the necessary microenvironmental conditions for the disseminated tumour cells’ survival’ [7]. For adaption to occur, they deploy mechanisms to modify the new microenvironment. To do so, along with the stroma cells, they establish a signalling network to promote their growth, to satisfy the metabolic demands to synthesise proangiogenic proteins in order to form new vascular networks and to facilitate their initial survival in the new ectopic location [8].
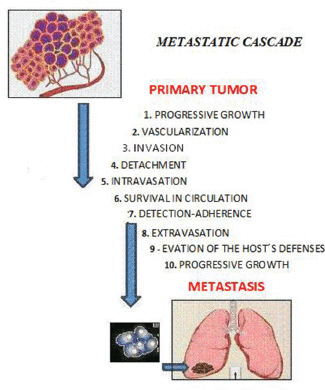
Figure 1. The sequential steps in the pathogenesis of metastasis are shown in the figure above, where each step is regulated by transitory and permanent changes in the DNA, the RNA, or by proteins. Also, most tumor cells fail to complete all of the steps, and the ‘few’ cells with metastatic ability ‘defeat’ the multiple mechanisms which impedes the formation of metastasis.
Metastasis is a largely ineffective process, leading it to be called ‘metastatic inefficiency’, since only 0.01% of the tumour cells that enter the bloodstream in animal models successfully form a secondary tumour [9]. With these models, it has been possible to choose cell populations with metastatic phenotypes and these cells in particular show increasing selectivity after each selection cycle for a specific organ [10]. Furthermore, the genome instability of neoplasms increases the likelihood of other cells developing skills that allow for the development of metastasis. Several studies have shown that an increase in metastatic ability is not the result of the adaption mechanisms of the tumour cells that allows for growth in a specific organ; rather, it is the gradual selection of a clone with different mutations to those observed in the primary tumour. The genome instability and heterogeneity of cancer cells is shown in the gains, losses, and chromosomal rearrangement of the tumours [11]. New technologies in parallel sequencing have allowed high-resolution genome analysis of cancer patients, making it possible to compare the primary tumour with regard to metastasis.
These studies have proven that there are inactive genes in the primary tumour, while they are active in metastasis, allowing for the activation of other oncogenes, confirming the concept of tumour heterogeneity [12]. Additionally, it has been shown that mutation patterns are shared in the metastasis of an organ, whereas metastasis in the different organs of a patient is different, establishing the hypothesis that metastasis results from clonal expansion where the subclones that have colonised an organ already have genetic alterations that allow specific adaption to the environment [13]. The period of time that passes between infiltrating the organ and colonising it is known as the latency period, where several tumour cells remain outside the cell cycle of secondary organs, while others are incapable of provoking the angiogenic changes necessary for tumour expansion [14].
It would appear that metastasis is a process determined by a complex network of interactions between metastatic cells and their microenvironment in the affected organs, making it necessary to update the contributions made for the knowledge of the different microenvironment elements involved in the formation of metastasis. It is necessary to focus on the main interactions established between the tumour cells and the microenvironment, from the primary tumour to reach the location where metastasis originates and develops. In all these phenomena, time has to also be considered as a factor of singular and paramount importance (Figure 2).
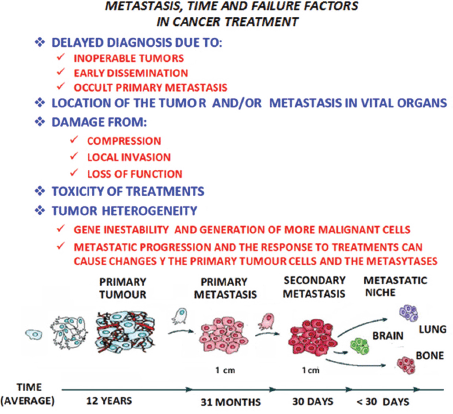
Figure 2. In metastatic progression, time and the failure factors indicated in the figure above, play a crucial role in the prognosis and treatment of the patient with cancer, as shown in the model of the linear progression of a tumor. In time, the cells increase in malignancy. Thus the concept of ‘metastasis of metastasis’ is used in reference to the metastasis becoming in the course of time more malignant, resulting in the death of the patient, as the illness cannot be controlled.
Components of the pre-metastastic niche
The metastatic niche plays an important role within the crucial factors that determine the success or failure of metastasis. A series of events take place in its formation, including: the modification of the extracellular matrix [ECM]; the restructuring of the vascular network; the participation of bone marrow cells; hypoxia and the expression of a wide variety of signalling molecules. Added to this is the participation of untransformed cells, such as fibroblasts and endothelial cells, as well as the deposition of molecules such as fibronectin, tenascin-c, and periostin [15]. Regarding particular tissue receptors, fibulin-5 reduces its levels so that matrix metallopeptidase 9 [MMP-9] restructures the matrix in metastasis in the liver and lungs, thus contributing to the formation of the metastatic niche [16]. Lysyl oxidase [LOX] actively participates in restructuring the extracellular matrix and the formation of the niche, so it has the ability to be linked to collagen and elastin. The expression of LOX is increased in tumour cells exposed to hypoxia conditions [17]. The development and consolidation of these investigations originated from the pioneering work made by Kaplan and co. [18], who used as many human tissue samples from cancer patients as Lewis lung carcinoma (LLC) and B16 cell lines from an animal model. They show that haematopoietic bone marrow-derived progenitor cells, which express vascular endothelial growth factor receptor 1 (VEGFR-1), form aggregated cells in pre-metastatic niches. The use of specific antibodies against VEGFR-1, as well as the elimination of these aggregated pre-metastatic cells, impedes the formation of metastasis. They also studied the adhesion and formation of aggregated cells after the implantation of the tumour cells, observing a growth in the expression of fibronectin and an increase in the dissemination of fibroblasts found in the primary tumour.
Furthermore, matrix metallopeptidases – particularly MMP-9 produced by bone marrow-derived progenitor cells – degrade the basal membrane, accelerating the extravasation of cells with VEGFR-1 phenotype in the niche. Along with fibronectin, associated with stroma cells, VEGFR-1 alters the local environment, activating integrin and chymosin to promote adhesion, survival, and growth in tumour cells. The primary tumour induces the production of MMP-9 in endothelial cells and macrophages in the lungs via a mechanism dependant on VEGFR-1/FLT-1 tyrosine kinase (TK) that promote metastasis. Additionally, they studied the expression of MMP-9 in healthy areas of the lungs in samples extracted from patients with tumours located in organs besides the lungs. In the lungs of patients with oesophageal cancer, melanoma skin cancer, ovarian cancer etcetera, a high expression of MMP-9 was found which suggests that primary tumours can stimulate the production of MMP-9 in pre-metastatic areas [19].
Types of cells
There are several types of cells that are important components of the metastatic niche, including heterogeneous populations in immune cells, endothelial cells, fibroblasts, macrophages, pericytes, and bone marrow-derived progenitor cells, which are described below:
Immune cells
They are represented by cells that actively participate in immunity processes, including macrophages, neutrophils, mast cells, myeloid-derived suppressor cells, dendritic cells, natural killer cells (NK) and more adaptive immune cells, such as T- and B-type lymphocytes. Immune cells that infiltrate the tumour, excluding NK cells, produce tumour cytokine promoters, such as tumour necrosis factor alpha [TNF-α] and interleukins IL-1β, IL-6, and IL-8, which increase signalling in pre-malignant cells. Likewise, this signalling not only stimulates tumour progression but also induces the production of cytokines by the tumour cells themselves [20].
Neutrophils
They are the most abundant human leukocytes as they are the first cells that gather at the infection site. Their degranulation releases lytic enzymes, as well as reactive oxygen species (ROS), hydrogen peroxide (H2O2), and hypochlorite (HOCL) with microbial potential [21]. Cytokines are also produced, such as TNF-α, IL-1β, IL-1Rα, IL-12, and VEGF; chemokines CXCL1, CXCL8, CXCL9, CXCL10, CCL3, and
CCL4 are involved in angiogenesis [22]. CXCL8 is produced in abundance by tumour cells, which once released into the microenvironment represent a potent chemo-attractant of neutrophils within the tumour. Furthermore, CXCL8 and other chemokines are associated with angiogenesis by the direct extracellular activation of CXCR2 [23], in particular, MMP-9 [24]. TNF-α, released in the tumour microenvironment, is linked with tumour progression inducing degranulation of the neutrophils, releasing VEGF and favouring angiogenesis with the production of CXCL8 and CXCL1 [25].
Dendritic cells
They represent a heterogeneous population made up of two types of cells: myeloids CD11c and CD123lo, and plasmacytoids CD11c− and CD123hi [26]. It has been shown that in various types of tumours, they present specific modifications in its stimulating ability with the consequent abnormal development of the differentiation of myeloid cells [27]. One of the mechanisms by which there is an abnormal differentiation of myeloid cells is the constitutive activation of the signal transducer and activator of transcription 3 [STAT3], which promotes continuing proliferation and the accumulation of immature myeloid cells, contributing to the suppression of the immune response before tumour angiogenesis [28]. Two pro-inflammatory molecules released by dendritic cells, TNF-α and osteopontin for instance, are associated with angiogenesis [29, 30]. Dendritic cells can secrete pro-angiogenic chemokines, such as CXCL1, CXCL2, CXCL3, CXCL5, CXCL8, and CCL2 [31]. In contrast, mature dendritic cells can inhibit angiogenesis by releasing cytokine IL-12 and chemokines CXCL9, CXCL10, and CCL21 [32].
Myeloid-derived suppressor cells (MDSC)
They represent an active role in the promotion of tumours and the evasion of the immune response. When they are immature cells, they seem to have monocyte/macrophage and granulocyte characteristics [33]. High levels of pro-inflammatory factors in the microenvironment of the tumour, such as GM-CSF, IL-1b, IL-6, and S-100, induce the recruitment and expansion of myeloid-derived suppressor cells, increasing pro-tumour activity [34]. Myeloid-derived suppressor cells also participate in the promotion of angiogenesis in the tumour through the release of soluble factors like MMP9 and VEGF. Experimental data suggest that these cells are also capable of being different from endothelial cells [35].
Natural killer cells
Natural killer (NK) cells are lymphocyte effectors of the innate immunity that could potentially control tumours by their cytotoxic activity. Like other types of cells, NK cells can infiltrate the mass, whose microenvironment is capable of affecting the functionality of these cells by a broad range of cytokines and soluble factors, either by inhibiting the cytotoxic function or promoting angiogenic phenotype. CD56bright and CD16- NK cells predominant in lung cancer exert lower cytotoxicity in K562 cells [36]. It has been reported that the invasion of NK cells in lung cancer (non-small-cell lung cancer [NSCLC]) produces high levels of VEGF, PIGF, and IL-8, inducing ‘ex vivo’ angiogenic activity [37]. The reduction in activity by NK cells is associated with the generation of the pre-metastatic niche and the efficiency of metastasis in murine models [38].
T cells
The inhibition of the flow of T-lymphocytes during angiogenesis and stroma restructuring represents a characteristic of the tumour microenvironment, giving way to alterations to its functionality. This is due to the activation and expansion of myeloid cells and soluble factors secreted by the tumour and inflammatory cells. The typical immunosuppressive tumour environment is characterised by a strong induction by CD4 , CD25 , FOXP3, and tumour-infiltrating regulatory T cells, and the activation of Th2 and Th17 [39, 40]. In ovarian cancer, hypoxia induces angiogenesis in humans and mice, where CD4 , CD25 and tumour-infiltrating regulatory T cells secrete high quantities of VEGFA and promote the dissemination of endothelial cells, both ‘in vitro’ and ‘in vivo’. In an ovarian tumour transplanted in a mouse, the depletion of tumour-infiltrating regulatory T cells correlates with a reduction in VEGFA, supporting the role of tumour-infiltrating regulatory T cells in angiogenesis in this type of tumour [41].
B cells
The stimulation of B cells culminates in the production of immunoglobulins [Ig] that participate in humoural immunity. They segregate a variety of cytokines such as interleukins IL-6 and IL-10, TNF-α, granulocyte-macrophage colony-stimulating factor (GM-CSF), and lymphotoxin (LT) that participate in humoural immunity [42]. In the development of solid tumours with similar characteristics to damaged tissues with immune dysfunction, such as chronic immune cell infiltration, tissue restructuring or angiogenesis, it is unsurprising that individuals with autoimmune diseases are at increased risk of developing cancer [43]. Complement proteins are associated with immunoglobulins and form circulating immune complexes (CIC), whose deposition in the parenchyma is due to defects in the vascular network, either by the tumour or by pathological angiogenesis, which starts a chain reaction [44]. In certain types of cancer, CIC levels in the tumour parenchyma are linked to an increase in tumour burden, indicating a poor prognosis [45]. In the K14-HPV16 mouse model, a carrier of squamous cell carcinoma (SCC), the suppression of B- and T-lymphocytes produced a decrease in angiogenesis and epithelial hyperproliferation. The transfer of B cells (B220 and CD19 ) in K14-HPV16 mice to K14-HPV16 mice (sic), deficient in T- and B-lymphocytes, restored malignant characteristics, such as hyperproliferation and angiogenesis. These data indicate that the activation of B cells is essential for the development of an epithelial neoplasm and that soluble mediators secreted by B cells are necessary to establish an inflammatory process that boosts tumour progression [46].
Mast cells
They represent a peculiar subtype of granulocytes that play a central role in the inflammatory process, participating in vascularisation during arthritis [47]. They have also been found to participate in the vascularisation of haematological malignancies, where they can integrate in the blood vessel wall by vascular mimicry [48]. The participation of mast cells in angiogenesis is associated with the production of various cytokines and chemokines [49]. Furthermore, proteases produced by mast cells promote angiogenesis [50]. B-triptase is a neutral protease that represents an abundant mediator found next to mast cell granules and plays an important role in inflammation, activating the release of protease by type 2 receptors that are directly involved in vascularisation [51].
Tumour-associated macrophages (TAM)
They act as regulators of tumourigenesis, either as residents or as derived from the spleen or bone marrow. Macrophages are typically considered as effector cells during immune defence but numerous studies have shown their role in tumour progression [52]. They are an important source of proteases, such as cysteine and cathepsin, that participate in tumour progression [53]. Tumour-associated macrophages have antagonistic functions between the homeostasis of normal tissues and tumorigenesis, which is why macrophages are functionally plastic and can alter their phenotype to suit different physiological conditions [54]. They can present a ‘M1’ phenotype that produces a type 1 pro-inflammatory cytokine that participates in antigen presentation, playing an antitumorigenic role, as well as a ‘M2’ phenotype that produces a type 2 cytokine that promotes the anti-inflammatory response and pro-tumorigenic function [55]. It has been suggested that in several environmental conditions, such as tumour hypoxia in a ‘M1’ to ‘M2’ transition, tumour-associated macrophages accumulate in hypoxic areas within the tumour, as well as endotheline-2 and VEGF. It should be noted that the accumulation of tumour-associated macrophages is linked with angiogenesis and the subsequent acquisition of invasive phenotypes [56]. Furthermore, a population of metastasis-associated macrophages (MAM) has been identified in a mouse model that promotes extravasation, dissemination and growth of breast cancer cells in the lungs. The inhibition of signalling by CCL2–CCR2 inhibits the accumulation of metastasis-associated macrophages and reduces metastasis [57]. Blood clotting plays an important role during metastasis, due to the blood-clotting proteins and tissue factor (TF) being linked to a poor prognosis for patients as they interfere with NK cells through the lysis of micrometastases. TF induces the formation of blood clots with platelets that stimulate the recruitment of bone marrow-derived macrophages, resulting in the survival of melanoma cells in the lungs. These blood clots recruit myeloid-derived suppressor cells in secondary niches, impeding the immunological rejection of the tumour [58].
Cancer-associated fibroblasts (CAF)
They are predominant cells in the connective tissue responsible for the elaboration of the extracellular matrix components and basal membrane, associated with the differentiation of epithelial cells and mediators of the immune response [59]. There are numerous cancer-associated fibroblasts in the tumour microenvironment, differing from normal fibroblasts. In mice, normal prostrate epithelial cells originate in intraepithelial tumours when coinjected with cancer-associated fibroblasts, but not when they are injected with normal fibroblasts [60]. Likewise, in breast cancer, cancer-associated fibroblasts stimulate the metastasis of malignant cells, while normal fibroblasts suppress metastasis [61]. This shows that cancer-associated fibroblasts make up a cell different to its normal counterpart. Moreover, their origin during the progression of the disease is unclear [62]; several studies suggest that they generate through the epithelial–mesenchymal transition of endothelial cells from blood vessels associated with tumours [63]. The mesenchymal–epithelial transition (MET) promotes cancer-associated fibroblast generation in tumours of epithelial origins; for instance, in breast cancer and prostate cancer, epithelial cells dedifferentiate to generate mesenchymal cells that express cancer-associated fibroblast markers [64]. Cancer-associated fibroblasts interact with tumour cells and additional components of the stroma through the production and secretion of various growth factors, cytokines, and chemokines. Once activated by the infiltration of immune cells, these fibroblasts produce pro-inflammatory chemokines such as CXCL1 and CXCL2 through the recruitment of tumour-associated macrophages in primary tumours [65], while CCL5 being secreted by these fibroblasts recruits tumour-infiltrating regulatory T cells by signalling through the CCR1 receptor expressed in these cells [66]. CCL5 is secreted by mesenchymal stem cells (MSC) that also act through the CCR5 receptor expressed by breast cancer cells, increasing the invasion and metastasis [67]. Moreover, CXCL12 and fibroblast growth factor receptor 2 (FGF-2), released by cancer-associated fibroblasts, stimulate neoangiogenesis by recruiting endothelial progenitor cells and vascular endothelial cells [68]. In mesenchymal–epithelial transition, tumour-associated fibroblasts are activated by TGF-β, PDGF, FGF, and proteases [69]. Once activated, cancer-associated fibroblasts secrete growth factors, including VEGF that induces vascular permeability and angiogenesis [70, 71].
Pericytes
They are specialised mesenchymal cells that are linked to smooth muscle, which act as support to endothelial cells and contribute both towards homoeostasis and the stabilisation, maturation and restructuring of capilliaries [72]. The intimate anatomical relationship between endothelial cells and pericytes suggests a stretched interaction between cell contacts by paracrine signalling. Platelet-derived growth factor B (PDGFB) is a family member of PDGF secreted by endothelial cells that joins with the tyrosine kinase receptor, PDGFR, expressed on the surface of pericytes. When PDGFB joins with PDGFR, dimerisation occurs and an intracellular signalling cascade that promotes cell proliferation and migration begins [73]. Angiopoietin-1 (Ang-1) is a soluble ligand produced by pericytes that joins with the tyrosine kinase receptor Tie-2, expressed by endothelial cells [74]. The interaction between Ang-1 and Tie-2 is fundamental for the maturation and stabilisation of the endothelium [75]. Transforming growth factor β (TGF-β) is a growth factor expressed by endothelial cells and pericytes during angiogenesis [76]. Vascularisation in tumours is chaotic and irregular, an instability that has been frequently attributed to a reduction in the number of pericytes [77]. The presence of pericytes can vary according to the type of tumour, considering that they increase in pancreatic cancer for example and decrease in glioblastoma, a notable fact when compared with the respective normal tissues. In reality, they are found in the majority of tumours, even though their association with the endothelium is abnormal [78]. Different studies have shown that they are essential in maintaining the tumour vascular network, as well as normal blood vessels, while the VEGF produced by the pericyte is necessary for the survival of endothelial cells in both contexts [79]. A hypothesis considers that the reduction of the number of pericytes in tumour vessels can increase intravasation of cancer cells, promoting its haematogenous dissemination [78]. In fact, it has shown the existence of an inverted link between the contents in pericytes of tumour vessels and the number of metastasis in colorectal cancer patients [80].
The composition of an inflammatory environment
An inflammatory environment may be created with the establishment of a tumour [81], as it has been observed that in many cases a pro-inflammatory environment is created which is composed of cytokines, chemokines, growth factors, activated stroma, metalloproteinases which degrade the extracellular matrix, and agents that cause damage to the DNA [82]. The inflammatory microenvironment introduced by the tumour cells affects the immune effector functions by means of immunosuppressor cells of the tumour-associated macrophage (TAM) type, immature Grl and Mac1 myeloid cells, T regulator lymphocytes, T reg, CD4 CD25 , natural killer T, NKT, etc. It is also possible that this occurs through the reduction of the number of dendritic cells, which are essential to initiating and maintaining an antitumour immune response [83]. The acute phase C-reactive protein or CRP and the A-amyloid or SAA proteins play a very important role in the induction of this inflammatory medium [84]. The SAA proteins are also chemotactic for other inflammatory cells such as the mastocytes and T lymphocytes [85], as well as in the induction of the expression of enzymes for the remodelling of the extracellular matrix [86] and in the production of inflammatory cytokines, such as TNF-α, that promote tumour growth [87]. The CD11b Gr1 myeloid suppressor cells inhibit the T and NK cells, protecting the tumour cells from immunological destruction [88]. The T regulator cells (Treg) are found in the tumour microenvironment and have several immunomodulator activities in cancer [89]. In normal physiological conditions, the Treg cells regulate the expansion and activation of the T and B lymphocytes, playing a critical role in the cytotoxic homoeostasis of the lymphocytes [90]. Based on the response to different environmental stimuli, the Treg cells have different effects on tumorigenesis, for example in breast tumours, which show that an increase in Treg cells is correlated with a lower survival [91], while in colorectal cancer, the Treg cells are associated with a higher survival [92]. Similar to the myeloid-derived suppressor cells or MDSCs, the Treg cells suppress the introduction of tumour-associated antigen and also interfere with the function of the cytotoxic T cells by means of inhibition through the release of cytolytic granules [93]. The S100A8 and S100A9 proteins, produced by primary tumours, induce the accumulation of haematopoietic progenitor cells and macrophages in pre-metastatic regions of the lung. In these regions, the serum amyloid A (SAA)3, induced by S100A8 and S100A9, acts as a regulating agent in the accumulation of the myeloid cells. In the lung, in the endothelial cells and macrophages, the toll-like4 (TLR4) receptor acts as a receptor for A(SAA)3 in the pre-metastatic phase, which also stimulates the signalling of NFKappa B, facilitating metastasis. This pro-inflammatory condition accelerates the migration of the primary tumour cells to the pre-metastatic niche in the lung [94]. In an animal model with metastasis in the lung, significant changes were observed in the vascular permeability that contributes to the establishment of the metastasis. The MD2 receptor represents a TLR4 coreceptor that creates regions of hyperpermeability by means of the overregulation of the CCR2 chemokine receptor. The CCR2-CCL2 system induces the secretion of permeability factors such as serum amyloid A3 and S100A8. This result suggests the possibility that the overregulation of CCR2 represents a marker for the regions of greater susceptibility to metastasis in lung cancer [95].
The epithelial–mesenchymal transition
Local invasion implies profound changes in the adhesion and the proteolytic and migratory properties of the tumour cells, which favours cellular disassociation, degradation of the extracellular matrix, and migration to adjacent tissues. In addition, the excessive proliferation of epithelial cells and angiogenesis are the markers of initiation and growth, which can be observed in a primary carcinoma [96]. During the progression of a carcinoma, the already differentiated tumour cells alter their genome, which confers an advantage on the cell in terms of growth. In later stages, the cells continue to change their genome and exhibit a non-differentiated phenotype frequently accompanied by a low expression of epithelial markers, which leads to a loss of intercellular junctions and epithelial polarity. These changes are often accompanied by an increase in the expression of mesenchymal markers, as well as the mobility of the cells, which gives them greater invasive capacity. The process by which the cells change from an epithelial phenotype to a mesenchymal phenotype is known as the epithelial–mesenchymal transition or EMT. This process can be defined as a cellular programme that permits the phenotype transition of the cell from epithelial to mesenchymal [97, 98].
The marginal zone of the tumour is an active and interactive region of great importance to the tumour microenvironment, where immune cells and stromal cells accumulate. In the case of the immature myeloid cells that accumulate in this region, they impede the differentiation of the antigen initiator cells, thus favouring the evasion of the tumour cells [99]. The macrophages are another type of principal cells that are found in the marginal zone and are recruited by the products secreted by the tumour cells [100]. Studies have demonstrated the importance of the stroma during the epithelial–mesenchymal transition in cancer by means of the presence or absence of TGF-β [101]. In teratocarcinomas, the accumulation of macrophages induces the epithelial–mesenchymal transition due to the TGF-β factor produced by the macrophages associated with the tumours [102].
The epithelial–mesenchymal transition can also be induced by TGF-β secreted by the platelets [103]. The macrophages also promote the invasion of tumour cells by means of the supply of migratory factors, such as EGF, which through the regulation of the production of fibrillar collagen, accelerate cellular motility and induces proteolytic activity for the remodelling of the extracellular matrix [104]. During the EMT transition, alterations occur in cell–cell adhesion, cell–substrate interaction, degradation of the extracellular matrix, and the reorganisation of the cytoskeleton [105]. EMT is fully achieved when degradation of the basal membrane occurs and a mesenchymatic cell can migrate, this acquiring invasive capacity, which permits metastatic dissemination. The activation of the EMT programme has been proposed as a critical mechanism for the acquisition of the malignant phenotype by the epithelial cells [106].
The cells that initiate the EMT transition are found on the invasive front of the primary tumours expressing mesenchymal markers that are capable of effecting intravasation, as they are transported through the circulation, into micro or macrometastatic forms [6]. In addition, the metastasis and the primary tumours are histologically similar, which can be interpreted as a reversible EMT that would permit migration and dissemination to different organs in the first place. Once the cells that have undergone EMT are in place, they would activate the opposite programme, the MET transition, which would permit them to establish secondary colonies by returning to epithelial morphology and re-acquiring the ability to grow and proliferate [107].
Transcriptional regulation of the epithelial–mesenchymal transition
A large number of molecular processes cooperate in the initiation and completion of the EMT transition, including the activation of transcription factors, the expression of specific surface proteins, the reorganisation and expression of proteins in the cytoskeleton, the production of enzymes that degrade the extracellular matrix, and the changes in the expression of miRNAs [108]. A key step in the EMT is the reduction in cell–cell adhesion by means of the transcriptional repression of the cadherins, components of the aherens junctions, such as occludin and claudin, components of tight junctions, to which is added the desmoplaquins, components of the desmosomes [109]. β-Catenin forms part of the adherens junctions, such that on breaking it migrates to the nucleus, where it functions as a cofactor for the Tcf or T-cell factor/lef transcription factors [110]. These then activate the transcription of genes such as c-myc, which increases cellular proliferation [111].
The expression of the intermediate filaments changes during the EMT with the substitution of keratin by vimentin [112], while the metalloproteinases increase during the EMT, participating in the loss of cell–cell junctions and the degradation of the basal membrane [113]. Several transcription factors that regulate the EMT inhibit apoptosis through activation of the MAPK and PI3K pathways [114]. During tumour progression, E-cadherin can be inactivated through the repression by means of the hypermethylation and deacetylation of the promoter through joining with the transcription receptors [115]. E-boxes, or short sequences of six nucleotides, -CACCTG or CAGGTG-, which determine specific expression in epithelial cells, were identified through analysis of the proximal promotor of E-cadherin in rats. The inactivation of these sequences activates the transcription of the E-cadherin in mesenchymal cells, which indicates the presence of repressors that silence the expression of this protein in non-epithelial cells [116]. The first repressors identified were the transcription factors with zinc fingers, Snail 1, and Snail 2 (slug) domains [117, 118], and the Zeb1 and Zeb2 transcription factors [119, 120], all of which are able to join with the E boxes of the cadherin promoter. Other repressors include the basic helix-loop-helix E12/E47 (TCF3) and Twist transcription factors [121, 122]. These repressors repress E-cadherin by means of the recruitment of some corepressors, such as for example, in the case of twist, activating the expression of other E-cadherin repressors. Furthermore, the overexpression of these factors in the epithelial cells not only produces the repression of E-cadherin, but also the reprogramming of the cell to a mesenchymal state. During the EMT, these repressors also repress other molecules from the adherens junctions and induce mesenchymal characteristics in a coordinated manner [123]. Also, the expression of Snail 1 induces the expression of fibronectin or vitronectin [124]. In the case of Twist, this regulator of the EMT induces the expression of the Akt kinase, a P13K effector and an important regulator in the survival pathways during the EMT [125].
The small non-coding RNa or microRNAs also act as regulators of the EMT transition, inhibiting the gene expression at the post-transcriptional level with the consequent reduction of the stability of the mRNAs, which are their target [126]. The constituents of the metastatic niche, as well as the remodelling of the ECM, have been associated with the induction of the EMT. Periostin or osteoblast-specific factor OSF-2 promotes its induction [127]. The EMT is induced by the metalloproteinases, MMPs that are activated in the metastatice niche [128]. Hypoxia also induces the activation of the EMT transition [129]. Other studies have demonstrated that the induction of EMT in the MCF10A cells with a high expression of SNAIL contributes to (a) resistance to antitumour drugs, (b) the acquisition of the mother cell phenotype through an increase in the expression of the CD44 /CD24− surface markers, (c) the capacity to form mammospheres [130]. In the case of the resistance to drugs developed by the mother cells, this represents one of the greatest challenges for chemotherapy against cancer [131]. This mainly affects tumour cells with rapid proliferation, while the mother cells grow slowly and have an effective resistance mechanism. This resistance to treatment leads to an increase in the rate of proliferation of the mother cells, which results in a recurrence of the cancer and metastasis [132].
Signalling pathways that activate the epithelial transition
The interaction of the tumour cells with the local microenvironment induces the autocrine or paracrine secretion of the growth factors, cytokines, and components of the extracellular matrix that can trigger the molecular programme for the EMT transition [133]. A large number of signalling pathways and growth factors have been associated with the EMT transition, such as the epidermal growth factor EGF, the fibroblastic growth factor FGF [134], and the hepatic growth factor or HGF [135]. The Wnt/β-catenin pathway is related to the EMT transition [136], while the TGF-β factor is another EMT inducer, as the signals activated by this factor inhibit some epithelial proteins, such as E-cadherin and keratin. They also activate the expression of mesenchymal proteins, such as fibronectin and vimentin, and in the case of TGF-β, it acts in the activation of the EMT through the Smad proteins [137]. Another pathway implicated in the induction of the EMT is that activated by NOTCH, which in turn is induced by hypoxia [138]. The NF-kβ factor acts as a regulator of the EMT, which is important for protection against apoptosis and in the induction of metastasis [139].
The metalloproteinases
The metalloproteinases or MMPs belong to a family of zinc-dependent endopeptidases that intervene both in the physiological processes of organogenesis and in the scar formation and in various pathological conditions, particularly cancer [140]. Although the first well-studied function of the metalloproteinases is the degradation of the ECM, it is currently believed that they play an important role in the processing of bioactive molecules such as growth factors, cytokines and chemokines, as well as their respective receptors [141]. The MMPs modulate the mediators of inflammation, such as the cytokines and chemokines, establishing the necessary gradients for chemotaxis of the inflammatory cells. They facilitate the migration of epithelial cells by interacting with these and the ECM proteins through proteolysis of the same matrix or the tissue adhesion proteins, such as E-cadherins [142]. In the vascular system, they influence the migration of tumour cells, the liberation of cytokines and growth factors linked to the cellular membrane, such as the transforming growth factor alpha TGF-α and the epidermal growth factor EGF [143]. In many malignant tumours, there is overexpression of ADAMs, desintegrin, and metalloproteinase, where their role in tumour growth and dissemination is related to their proteolytic activity [144]. Several MMPs and MT-MMPs have an important activating action on other pro-MMPs, while they are activated by other proteases. These processes usually take place in the extracellular space, but there is a group of proteases, the MT-MMPs, MMP-11, MMP-23, and MMP-28, which activate within the cell by means of a furin-type proprotein convertase [145]. The MMPs also act as signalling molecules and can then modulate other cell signalling molecules. The platelet-derived growth factor PDGF produces an increase in the expression of MMP-1, which acts in conjunction with TGF-β, producing an overexpression of MMP-3 and TIMP-1 [146]. The epidermal growth factor EGF induces the expression of MMP-1 [147], and the vascular endothelial growth factor VEGF and fibroblast growth factor FGF-2 are angiogenic factors that can induce the expression of the MMP proteases, facilitating metastatic dissemination [148]. The tumour necrosis factor alpha TGF-α is a pro-inflammatory cytokine liberated by macrophages, T lypmhocytes, and mastocytes that induce the overexpression of some MMPs such as MMP-2, MMP-3, MMP-7, and MMP-9 in the tumour microenvironment, which increases the invasive capacity of the malignant cells [149].
The metalloproteinases and the progression of cancer
The MMPs are the principal mediators in the changes observed in the tumour microenvironment during the progression of cancer [150], which play a fundamental role in tumour growth through degradation of the connective tissue and the components of the basal membrane, in addition to activating growth factors, surface receptors for adhesion molecules and for chemokines [151]. This interaction with the components of the ECM alters the cellular response to the microenvironment, making the tumour cells less adherent so they have more potential to migrate and produce metastasis [152]. During carcinogenesis, the tumour cells interact with the growth factors, the cytokines, and other cells, such as endothelial cells, fibroblasts, macrophages, mastocytes, and pericites, that are present in the tumour microenvironment [153]. All of this demonstrates that the remodelling of the ECM is an active event during tumour progression that leads to the formation of a niche for the survival and proliferation of the tumour cells [154].
The MMPs can play different roles during the progression of the cancer, depending on the stage of the tumour. In early stages, the proteolysis of MMPs 3 and 7, which link growth factors, contribute to cellular proliferation, but later, the cleavage of E-cadherin and CD44 activates the motility of the tumour cells facilitating metastasis [155]. In contrast, MMP-8 has a protective effect as it diminishes the metastatic potential of breast cancer cells [156], but in the case of the overexpression of MMPs 2 and 9, which indicates an unfavourable prognosis as it degrades type 4 collagen, located in the basal membranes, and induces the expression of angiogenic factors [157].
The local invasion of the tumours depends on the degradation of the proteins of the basal membranes, such as type 4 or 5 collagen, and the proteolysis of type 1, 2, or 3 interstitial collagen present in the connective tissue that surrounds the tumour cells [158]. In addition, the MMPs intervene in angiogenesis, promoting the migration of the endothelial cells, liberating VEGF and other proangiogenic factors of the ECM, such as FGF-2 and TGFβ, which also favour the proliferation and migration of these cells [159, 154]. In animal models, it has been demonstrated that the MMPs regulate the formation and maturation of new blood vessels through the control they exercise on the growth factors and cytokines, which act on the recruitment of pericytes [160]. Some MMPs can have an inhibiting effect on angiogenesis; for example, the hydrolysis of plasminogen creates fragments of angiostatin and the proteolysis of XVIII collagen produces endostatin [161]. The transcription of the MMPs is induced by inflammatory cytokines, such as IL-1, IL-6, TNF-α, and growth factors such as EGF, HGF, and TGF-β, giving them a preponderant role in the chronic inflammation which is present in the tumour microenvironment. Other factors, such as TGF-α and IL-4 inhibit their expression, and can be considered as therapeutic targets for cancer [162].
Angiogenesis
Angiogenesis is a crucial mechanism in the development of cancer due to the growing need for oxygen and nutrients, as the lack of these will lead to the latency and death of the tumour cells [163]. Tumour angiogenesis depends on both the angiogenic growth factors, with stimulants such as angiogenin, angiopoietin-1, cyclo-oxygenase, hepatocyte growth factor, and tumour growth factor produced by the tumour cells and those of the host, and the antiangiogenic factors, which are inhibitors and include angiopoietin-2, angiostatin, interferon-α/β endostatin, and vasostatin, which keep the existing blood vessels in a state of quiescence [164].
The vascularisation of the tumour requires the participation of many types of cells in the tumour microenvironment, including the vascular endothelial cells, the pericytes, and the bone marrow precursor cells, whose participation is regulated by hypoxia [165, 166]. Added to these are the tumour-associated macrophages, the tumour-associated fibroblasts, and the mesenchymal mother cells, which also contribute to the vascularisation of the tumour through the liberation of pro-angiogenic signals in the tumour microenvironment. In patients with advanced, vascularised breast cancer, a high mobilisation of mesenchymal mother cells was found in the circulation, which indicates that these cells have an active participation in tumour progression [167, 168]. Lymphangiogenesis is another route for tumour vascularisation, as the cancer cells can also be disseminated through the lymph vessels [169]. In human cervical cancer, the activated macrophages can produce the VEGF-C and VEGF-D factors, which correlate with lymphangiogenesis [170].
In order to initiate neo-vascularisation, an avascular tumour must acquire an angiogenic phenotype that permits it to ‘turn on’ the angiogenic interruptor [171]. Obviously, the mechanism through which the angiogenic interruptor is ‘connected’ must involve partial oxygen pressure sensors that are sufficiently sensitive to rapidly detect the hypoxia produced in the interior of the solid avascular tumour [172]. The response to hypoxia is the induction of the gene that codes the VEGF protein, considered the most potent angiogenic inducer, while it is also very likely that other factors exist, which can play a role in the response of the tumour to hypoxia [173]. It is accepted that the angiogenic interruptor is ‘turned off’, while the effect of the pro-angiogenic molecules is suspended by the antiangiogenic molecules, and the interruptor turns on when the net balance shifts in favour of angiogenesis. The net angiogenic activity of a tumour is the result of the disequilibrium between stimulating and inhibiting signals [174].
Tumour latency and progression
Tumours can remain in a latent state for years when there is an equilibrium between proliferation and apoptosis, a phenomenon that can be defined ‘as a state of temporal latency of the arrest of tumour growth’ [175]. This condition can be divided into three categories:
(a) Cellular latency
Structural components of the niche can promote the survival of the tumour cells while keeping them in a state of latency [176]. Such is the case of the cytoskeleton, which can reactivate these cells, which suggests that the rigidity of the extracellular matrix presents the property of promoting the exit from latency. In an in vitro model using cells from a hepatocellular carcinoma, the increase in rigidity of the microenvironment was linked to the TGF-ß factor through induction of the D1 and D3 cyclins [177], showing that less rigid environments favour the latency of tumour cells. In a breast cancer model, it was shown that TGF-ß1 induced fibrosis through deposition of type 1 collagen in the lung, which favoured the escape from latency [178]. In breast cancer, the fibroblasts associated with the cancer can interact with proteins in the extracellular matrix to modulate the intracellular adhesions, cellular contractibility, and forces within the tumour microenvironment. It has been demonstrated that caveolin-1, or Cav-1, produced by the fibroblasts promotes the rigidity of the tumour microenvironment through the activation of GTPasa, thus stimulating tumour progression [179]. The induction of the EMT within the niche causes the tumour cells to enter into a state of latency due to the increase in the levels of p16INK4a [180], the repression of cyclin D [181] and a sustained expression of twist [182]. The latency confers resistance to tumour cells against the action of antitumour agents, whether due to arrest of the cell cycle or a slow proliferation which makes the action of the antitumour agents ineffective [183]. On the other hand, the latent cells must be reactivated in order to grow and form a metastasis. Once the niche has matured, some of its components may act to liberate the cells from latency. Changes in the cytokines can liberate the latent cells, induced by the TCD4 cells [184]. Also, the remodelling of the extracellular matrix by type 1 collagen has been implicated in the liberation from latency through integrin signalling mediated by FAK [185]. The uPAR urokinase receptor activates the β1 integrin, which on interacting with fibronectin, permits the liberation of the tumour cell from latency [186].
(b) Angiogenic latency
As with normal tissues, the tumours provide nutrients to the cells, such that an inefficient vascularisation causes the tumour mass to remain constant due to an equilibrium between the cells that are dividing and those which are dying. This is the reason why tumour cells, when forming a micrometastasis, must vascularise it in order to survive, as if this does not occur, it can disappear or enter into a state of latency [187], a condition in which it will remain until genetic, epigenetic, and microenvironmental signals can activate angiogenesis. In a study of immunodeficient mice who carried a liposarcoma that was able to remain latent for more than 90 days, high levels of thrombospondin (TSP) and angiomotin were demonstrated [188]. Thrombospondin or TSP is a glycoprotein of the cellular matrix which in physiological conditions is segregated by the fibroblasts and other cells, such as the endothelial cells [189]. On the other hand, TSP was found in latent breast cancer tumour cells that were in contact with the microvasculature of the organs in which they would have metastasised. In a three-dimensional in vitro model, the endothelial cells that are part of the vascular system produce the TSP, which acts as a suppressor of angiogenesis and causes the tumour cells to remain in a state of latency. This can explain the effect of TSP on the latency of tumour cells [190]. TSP is a suppressor protein of angiogenesis, however, with the formation of new vessels the endothelial cells produce TGF-ß1 and periostin (POSTN), which will permit the latent cells to initiate their proliferation [191]. The overexpression of Notch-delta 4 (DLL4) in endothelial cells can promote the exit of T-ALL cells from a latent state through joining with the Notch 3 receptor. This overexpression of DLL4 can be induced by the vascular endothelial growth factor VEGF, with the resulting exit from the latent state [192].
(c) Immunologic latency
A critical step in tumour progression is the evasion and suppression of the immune system of the host [193], which can be achieved through the inhibition of immune effector cells or through the stimulation of immunosuppressor cells. One of the most common mechanisms of immune evasion in patients is due to the activity of the myeloid-derived suppressor cells or MDSCs, which are defined as immature immunosuppressant myeloid cells that maintain the homoeostasis of normal tissue in response to adverse situations such as infections, post-traumatic stress, etc [194]. During tumorigenesis, the MDSC cells infiltrate the tumour promoting its vascularisation [195] and disturbing the immune mechanisms such as the production of antigens by dendritic cells or DCs [196], the activation of T cells [197], the polarisation of M1 macrophages [198] and the inhibition of the cytotoxic NK cells [199]. The MDSC cells promote tumour progression, which has been demonstrated in animal models, and is supported by the elevated number of these cells which is found in patients with cancer, which correlates with an advanced stage of the disease and treatment failure [200].
Stroma and growth of the tumour cells: pre-clinical studies
As we have seen, in a tumour the different cells of the stroma can interact directly with each other through direct contact or through paracrine signalling. In particular case of the tumour cells, these can induce changes in the cells of the stroma and the extracellular matrix through several mechanisms. They can also alter the stroma through cell–cell contact, modify the extracellular matrix, or liberate soluble factors. They often secrete cytokines, chemocytokines, growth factors, metalloproteinases, and inflammatory mediators that can promote invasivity. However, the mechanisms through which the stroma cells facilitate the growth of tumour cells are not known, and further studies are needed to understand the mechanisms implicated between the cells and the tumour microenvironment in order to design new strategies against cancer and improve existing treatments. Preclinical studies have tested treatments blocking the mechanisms that the tumour cells develop to evade the immune response. Ipilimumab, an antibody that activates the T cells, promoting antitumour activity, was tested on patients with metastatic melanoma, and increased their survival in comparison to patients not treated with this antibody [201]. The CD40 antibody reverses immune suppression activating the antigen-producing cells and promoting the antitumour response of the T cells [202]. An elevated expression of programmed cell death protein 1, PD-1, is produced with cancer.
It has been demonstrated that Pembrolizumab or MK-3475 blocks PD-1 and its ligand PD-L1 expressed in tumour cells in the tumour microenvironment, both in melanomas and in other types of tumours [203]. Dasatinib, an Src gene inhibitor, makes it difficult for melanoma tumour cells to survive [204] as CSF-1R, a regulator or the macrophages associated with tumours, inhibits apoptosis, migration, and invasion in the breast cells of canines [205]. Nintedanib is a simultaneous inhibitor of VEGFR, PDGFR, and FGR, which shows antiangiogenic and antineoplasic inhibitory activity in lung cancer, impeding the growth of tumour cells [206].
In addition, products have been used, which impede the degradation of bone to avoid its colonisation by tumour cells, as is the case with prostate cancer. Micro RNA, miR-34a, acts as a suppressor of osteoclastogenesis, bone resorption, and the formation of a metastatic niche, impeding the establishment of metastatic cells [207]. Resveratrol inhibits the EMT transition in the cellular line of LoVo colon cancer through the inhibition of the TGF-β1/Smads pathway mediated by the expression of Snail/E-cadherin, impeding invasion and metastasis [208]. Curcumin inhibits the EMT transition and induces apoptosis in PANC-1 pancreatic cells through the Shh-GLI1 pathway [209].
Conclusion
Cancer is not only the transformation of individual cells into a state of cellular proliferation, but a disruption of the forms in which the tissues regulate their processes and affect the systemic interactions with the affected organism. Currently, the fundamental treatments against cancer continue to be surgery, radiation therapy, and chemotherapy, which usually destroys the primary tumour, but whose action is very limited against metastasis. This is why it is necessary to continue investigating to find new prognostic markers and new therapeutic targets for metastasis before it occurs, as the early detection of these markers could determine which cases require treatment and avoid it in those patients without a risk of metastasis. Thus, for example, the monitoring of growth factors and cytokines in the blood which may induce the formation of the premetastatic niche would be fundamental. At the same time, determining the blood levels of components of the metastatic niche such as the VEGFR1 protein circulating or interfering with the formation of inflammatory components such as type CD11b myeloid cells is indispensable for this purpose.
Genetic, cellular biology, and molecular studies, as well as those of the internal and external environmental contexts, indicate that tumour growth is not only determined by its cells, but also by the tumour microenvironment and the entire context in which the organism functions. In this way, the progression of cancer is the result of a very complex relationship between the different malignant and non-malignant cell types, components of the stroma, and the entire body of the organism.
Due to the implication of metastasis in mortality due to cancer, it is also necessary to search for new ways to integrate the two focuses which dominate the current science: the reductionist vision and the systemic vision sustained by the science of complexity.
References
1. Lugassy C and Escande JP (1997) The haematogenous theory of metastasis: récamier did not propose it Virchows Arch 431(5) 371 DOI: 10.1007/s004280050113
2. Valastyan S and Weinberg RA (2011) Tumor metastasis: molecular insights and evolving paradigms Cell 147 275–92 DOI: 10.1016/j.cell.2011.09.024 PMID: 22000009 PMCID: 3261217
3. Arvelo F and Poupon MF (2001) Aspectos moleculares y celulares de la metástasis cancerosa Acta Cient Venez 52 304–12
4. Langley RR and Fidler IJ (2007) Tumor cell-organ microenvironment interactions in the pathogenesis of cancer metastasis Endocr Rev 28 297–321 PMID: 17409287
5. Talmadge JE, Benedict K and Madsen J et al (1984) Development of biological diversity and susceptibility to chemotherapy in murine cancer metastases Cancer Res 44 3801–5 PMID: 6744297
6. Arvelo F (2013) Micrometástasis: estrategias para su detección Invest Clínic 54 206–25
7. Psaila B ans Lyden D (2009) The metastatic niche: adapting the foreign soil Nat Rev Cancer 9 285–93 DOI: 10.1038/nrc2621 PMID: 19308068 PMCID: 3682494
8. Folkman J (1971) Tumor angiogenesis: therapeutic implications N Engl J Med 285 1182–6 DOI: 10.1056/NEJM197111182852108 PMID: 4938153
9. Fidler IJ (1970) Metastasis: quantitative analysis of distribution and fate of tumor emboli labeled with 125 I-5-iodo-2’-deoxyuridine J Natl Cancer Inst 45 773–82 PMID: 5513503
10. Hill RP, Young SD and Cillo C et al (1986) Metastatic cell phenotypes: quantitative studies using the expèrimental metástasis assay Cancer Rev 5 118–51
11. Ruddon RW (1995) Cancer biology Oxford University Press New York
12. Vizoso M, Ferreira HJ and Lopez-Serra P et al (2015) Epigenetic activation of a cryptic TBC1D16 transcript enhances melanoma progression by targeting EGFR Nat Med DOI: 10.1038/nm.3863 PMID: 26030178
13. De Vita VT, Hellman S and Rosemberg SA (2004) Cancer, principles & practice of oncology Lippincott & Wilkins New York
14. Ghajar CM, Peinado H and Mori H et al (2013) The perivascular niche regulates breast tumour dormancy Nat Cell Biol 15 807–17 DOI: 10.1038/ncb2767 PMID: 23728425 PMCID: 3826912
15. Oskarsson T, Acharyya S and Zhang XH et al (2011) Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs Nature Med 17 867–74 DOI: 10.1038/nm.2379 PMID: 21706029 PMCID: 4020577
16. Moller HD, Ralfkjaer U and Cremers N et al (2011) Role of fibulin-5 in metastatic organ colonization Mol Cancer Res 9 553–63 DOI: 10.1158/1541-7786.MCR-11-0093 PMID: 21454378
17. Wong CC, Gilkes DM and Zhang H et al (2011) Hypoxia-inducible factor 1 is a master regulator of breast cancer metastatic niche formation Proc Natl Acad Sci USA 108 16369–74 DOI: 10.1073/pnas.1113483108 PMID: 21911388 PMCID: 3182724
18. Kaplan RN, Riba R and Zacharoulis S et al (2005) VEGFR-1 positive haemapoietec bone marrow progenitors initiate the pre-metastatic niche Nature 438 820–7 DOI: 10.1038/nature04186 PMID: 16341007 PMCID: 2945882
19. Kaplan RN, Rafiis S and Lyden D (2006) Preparing the “soil” the premetastatic niche Cancer Res 66 11089–93 DOI: 10.1158/0008-5472.CAN-06-2407 PMID: 17145848 PMCID: 2952469
20. Grivennikov SI, Greten FR and Karin M (2010) Immunity, inflammation, and cancer Cell 140 883–99 DOI: 10.1016/j.cell.2010.01.025 PMID: 20303878 PMCID: 2866629
21. Borregaard N and Cowland JB (1997) Granules of the human neutrophilic polymorphonuclear leukocyte Blood 89 3503–21 PMID: 9160655
22. Scapini P, Lapinet-Vera JA and Gasperini S et al (2000) The neutrophil as a cellular source of chemokines Immunol Rev 177 195–203 DOI: 10.1034/j.1600-065X.2000.17706.x
23. Strieter RM, Burdick MD and Gomperts BN et al (2005) CXC chemokines in angiogenesis Cytokine Growth Factor Rev 16 593–609 DOI: 10.1016/j.cytogfr.2005.04.007 PMID: 16046180
24. Hanahan D and Coussens LM (2012) Accessories to the crime: functions of cells recruited to the tumor microenvironment Cancer Cell 21 309–22 DOI: 10.1016/j.ccr.2012.02.022 PMID: 22439926
25. Balkwill F (2006) TNF-alpha in promotion and progression of cancer Cancer Metastasis Rev 25 409–16 DOI: 10.1007/s10555-006-9005-3 PMID: 16951987
26. Liu K and Nussenzweig MC (2010) Origin and development of dendritic cells Immunol Rev 234 45–54 DOI: 10.1111/j.0105-2896.2009.00879.x PMID: 20193011
27. Gabrilovich D (2004) Mechanisms and functional significance of tumour-induced dendritic-cell defects Nat Rev Immunol 4 941–52 DOI: 10.1038/nri1498 PMID: 15573129
28. Yu H, Pardoll D and Jove R (2009) STATs in cáncer inflammation and immunity: a leading role for STAT3 Nat Rev Cancer 9 798–809 DOI: 10.1038/nrc2734 PMID: 19851315
29. Braumuller H, Wieder T and Brenner E et al (2013) T-helper-1-cell cytokines drive cancer in to senescence Nature 494 361–365 DOI: 10.1038/nature11824
30. Shinohara ML, Lu L and Bu J et al (2006) Osteopontin expression is essential for interferon-alpha production by plasmacytoid dendritic cells Nat Immunol 7 498–506 DOI: 10.1038/ni1327 PMID: 16604075 PMCID: 3725256
31. Scimone ML, Lutzky VP and Zittermann SI et al (2005) Migration of polymorphonuclear leucocytes is influenced by dendritic cells Immunol 114 375–85 DOI: 10.1111/j.1365-2567.2005.02104.x
32. Albini A, Brigati C and Ventura A et al (2009) Angiostatin anti-angiogenesis requires IL-12: the innate immune system as a key target J Transl Med 7 5–13 DOI: 10.1186/1479-5876-7-5 PMID: 19144161 PMCID: 2630934
33. Peranzoni E, Zilio S and Marigo I et al (2010) Myeloid-derived suppressor cell heterogeneity and subset definition Curr Opin Immunol 22 238–44 DOI: 10.1016/j.coi.2010.01.021 PMID: 20171075
34. Bunt SK, Sinha P and Clements VK et al (2006) Inflammation induces myeloid-derived suppressor cells that facilitate tumor progression J Immunol 176 284–290 DOI: 10.4049/jimmunol.176.1.284
35. Murdoch C, Muthana M and Coffelt SB et al (2008) The role of myeloid cells in the promotion of tumour angiogenesis Nat Rev Cancer 8 618–31 DOI: 10.1038/nrc2444 PMID: 18633355
36. Platonova S, Cherfils-Vicini J and Damotte D et al (2011) Profound coordinated alterations of intratumoral NK cell phenotype and function in lung carcinoma Cancer Res 71 5412–22 DOI: 10.1158/0008-5472.CAN-10-4179 PMID: 21708957
37. Bruno A, Focaccetti C and Pagani A et al (2013) The proangiogenic phenotype of natural killer cells in patients with non-small cell lung cancer Neoplasia 15 133–42 DOI: 10.1593/neo.121758 PMID: 23441128 PMCID: 3579316
38. Sceneay J, Chow MT and Chen A et al (2012) Primary tumor hipoxia recruits CD11b /Ly6Cmed/Ly6G immune suppressor cells and compromises NK cell cytotoxicity in the premetastatic niche Cancer Res 72(16) 3906–11 DOI: 10.1158/0008-5472.CAN-11-3873 PMID: 22751463
39. Motz GT and Coukos G (2011) The parallel lives of angiogenesis and immunosuppression: cáncer and other tales Nat Rev Immunol 11 702–11 DOI: 10.1038/nri3064 PMID: 21941296
40. Hoechst B, Gamrekelashvili J and Manns MP et al (2011) Plasticity of human Th17 cells and iTregs is orchestrated by different subsets of myeloid cells Blood 117 6532–41 DOI: 10.1182/blood-2010-11-317321 PMID: 21493801
41. Facciabene A, Peng X and Hagemann IS et al (2011) Tumour hipoxia promotes tolerance and angiogenesis via CCL28 and T (reg) cells Nature 475 226–30 DOI: 10.1038/nature10169 PMID: 21753853
42. Pillai S, Mattoo H and Cariappa A (2011) B cells and autoimmunity Curr Opin Immunol 23 721–731 DOI: 10.1016/j.coi.2011.10.007 PMID: 22119110 PMCID: 3268048
43. Bernatsky S, Ramsey-Goldman R and Clarke A (2006) Malignancy and autoimmunity Curr Opin Rheumatol 18 129–34 DOI: 10.1097/01.bor.0000209423.39033.94 PMID: 16462517
44. Gunderson AJ and Coussens LM (2013) B cells and their mediators as targets for therapy in solid tumors Exp Cell Res 319 1644–9 DOI: 10.1016/j.yexcr.2013.03.005 PMID: 23499742 PMCID: 3743954
45. Lu H, Goodell V and Disis ML (2008) Humoral immunity directed against tumor-associated antigens as potential biomarkers for the early diagnosis of cancer J Proteome Res 7 1388–94 DOI: 10.1021/pr700818f PMID: 18311901
46. De Visser KE, Korets LV and Coussens LM (2005) De novo carcinogenesis promoted by chronic inflammation is B lymphocyte dependent Cancer Cell 7 411–23 DOI: 10.1016/j.ccr.2005.04.014 PMID: 15894262
47. Mauri C and Bosma A (2012) Immune regulatory function of B cells Annu Rev Immunol 30 221–41 DOI: 10.1146/annurev-immunol-020711-074934 PMID: 22224776
48. Nico B, Mangieri D, Crivellato E and Vacca A et al (2008) Mast cells contribute to vasculogenic mimicry in multiple myeloma Stem Cells Dev 17 19–22 DOI: 10.1089/scd.2007.0132 PMID: 18205547
49. Marichal T, Tsai M and Galli SJ (2013) Mast cells: potential positive and negative roles in tumor biology Cancer Immunol Res 1 269–79 DOI: 10.1158/2326-6066.CIR-13-0119
50. de Souza DA Jr, Toso VD and Campos MR et al (2012) Expression of mast cell proteases correlates with mast cell maturation and angiogenesis during tumor progression PLoS One 7(7) e40790 Epub 2012 Jul 18 DOI: 10.1371/journal.pone.0040790 PMID: 22815822 PMCID: 3399855
51. Ammendola M, Sacco R and Sammarco G et al (2014) Correlation between serum tryptase, mast cells positive to tryptase and microvascular density in colorectal cancer patients: possible biological-clinical significance PLoS One 9(6) e99512 DOI: 10.1371/journal.pone.0099512
52. Qian BZ and Pollard JW (2010) Macrophage diversity enhances tumor progression and metastasis Cell 141 39–51 DOI: 10.1016/j.cell.2010.03.014 PMID: 20371344
53. Shree T, Olson OC and Elie BT et al (2011) Macrophages and cathepsin proteases blunt chemotherapeutic response in breast cancer Genes & dev 25 2465–79 DOI: 10.1101/gad.180331.111
54. Mosser DM and Edwards JP (2008) Exploring the full spectrum of macrophage activation Nat Rev Immunol 8 958–69 DOI: 10.1038/nri2448 PMID: 19029990 PMCID: 2724991
55. Biswas SK and Mantovani A (2010) Macrophage plasticity and interaction with lymphocyte subsets: cáncer as a paradigm Nat Immunol 11 889–96 DOI: 10.1038/ni.1937 PMID: 20856220
56. Escribese MM, Casas M and Corbi AL (2012) Influence of low oxygen tensions on macrophage polarization Immunobiol 217 1233–40 DOI: 10.1016/j.imbio.2012.07.002
57. Qian BZ, Li J and Zhang H et al (2011) CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis Nature 475 222–5 DOI: 10.1038/nature10138 PMID: 21654748 PMCID: 3208506
58. Gil-Bernabe AM, Ferjancic S and Tlalka M et al (2012) Recruitment of monocytes/macrophages by tissue factor-mediated coagulation is essential for metastatic cell survival and premetastatic niche establishment in mice Blood 119 3164–75 DOI: 10.1182/blood-2011-08-376426 PMID: 22327225
59. Kalluri R and Zeisberg M (2006) Fibroblasts in cancer Nat Rev Cancer 6 392–401 DOI: 10.1038/nrc1877 PMID: 16572188
60. Olumi AF, Grossfeld GD and Hayward SW et al (1999) Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium Cancer Res 59 5002–11 PMID: 10519415
61. Dumont N, Liu B and Defilippis RA et al (2013) Breast fibroblasts modulate early dissemination, tumorigenesis, and metastasis through alteration of extracellular matrix characteristics Neoplasia 15 249–62 DOI: 10.1593/neo.121950 PMID: 23479504 PMCID: 3593149
62. Marsh T, Pietras K and McAllister SS (2013) Fibroblasts as architects of cancer pathogenesis Biochimica et Biophysica Acta 1832 1070–8 DOI: 10.1016/j.bbadis.2012.10.013 PMCID: 3775582
63. Potenta S, Zeisberg E and Kalluri R (2008) The role of endothelial-to-mesenchymal transition in cancer progression Br J Cancer 99 1375–9 DOI: 10.1038/sj.bjc.6604662 PMID: 18797460 PMCID: 2579683
64. Orr B, Riddick AC and Stewart GD et al (2012) Identification of stromally expressed molecules in the prostate by tag-profiling of cancer-associated fibroblasts, normal fibroblasts and fetal prostate Oncogene 31 1130–1142 DOI: 10.1038/onc.2011.312 PMCID: 3307063
65. Erez N, Truitt M and Olson P et al (2010) Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-kappaB-dependent manner Cancer Cell 17 135–47 DOI: 10.1016/j.ccr.2009.12.041 PMID: 20138012
66. Tan W, Zhang W and Strasner A et al (2011) Tumour-infiltrating regulatory T cells stimulate mammary cancer metastasis through RANKL-RANK signalling Nature 470 548–53 DOI: 10.1038/nature09707 PMID: 21326202 PMCID: 3166217
67. Karnoub AE, Dash AB and Vo AP et al (2007) Mesenchymal stem cells within tumour stroma promote breast cancer metastasis Nature 449 557–63 DOI: 10.1038/nature06188 PMID: 17914389
68. Orimo A, Gupta PB and Sgroi DC et al (2005) Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion Cell 121 335–48 DOI: 10.1016/j.cell.2005.02.034 PMID: 15882617
69. Marsh T, Pietras K and McAllister SS (2013) Fibroblasts as architects of cancer pathogenesis Biochimica et Biophysica Acta 1832 1070–8 DOI: 10.1016/j.bbadis.2012.10.013 PMCID: 3775582
70. Fukumura D, Xavier R and Sugiura T et al (1998) Tumor induction of VEGF promoter activity in stromal cells Cell 94 715–25 DOI: 10.1016/S0092-8674(00)81731-6 PMID: 9753319
71. Chowdhury R, Webber JP and Gurney M et al (2015) Cancer exosomes trigger mesenchymal stem cell differentiation into pro-angiogenic and pro-invasive myofibroblasts Oncotarget 6(2) 715–31 DOI: 10.18632/oncotarget.2711 PMID: 25596732 PMCID: 4359250
72. Dore-Duffy P and Cleary K (2011) Morphology and properties of pericytes Methods Mol Biol 686 49–68 DOI: 10.1007/978-1-60761-938-3_2
73. Heldin CH and Westermark B (1999) Mechanism of action and in vivo role of platelet-derived growth factor Physiol Rev 79 12831316
74. Sundberg C, Kowanetz M and Brown LF et al (2002) Stable expression of angiopoietin-1 and other markers by cultured pericytes: phenotypic similarities to a subpopulation of cells in maturing vessels during later stages of angiogenesis in vivo Lab Invest 82 387–401 DOI: 10.1038/labinvest.3780433 PMID: 11950897
75. Falcón BL, Hashizume H and Koumoutsakos P et al (2009) Contrasting actions of selective inhibitors of angiopoietin-1 and angiopoietin-2 on the normalization of tumor blood vessels Am J Pathol 175 2159–70 DOI: 10.2353/ajpath.2009.090391 PMID: 19815705 PMCID: 2774078
76. Goumans MJ, Valdimarsdottir G and Itoh S et al (2002) Balancing the activation state of the endothelium via two distinct TGF-beta type I receptors EMBO J 21 1743–53 DOI: 10.1093/emboj/21.7.1743 PMID: 11927558 PMCID: 125949
77. Barlow KD, Sanders AM and Soker S et al (2013) Pericytes on the tumor vasculature: jekyll or hyde? Cancer Micro environ 6 1–17 DOI: 10.1007/s12307-012-0102-2
78. Raza A, Franklin MJ and Dudek AZ (2010) Pericytes and vessel maturation during tumor angiogenesis and metastasis Am J Hematol 85 593–8 DOI: 10.1002/ajh.21745 PMID: 20540157
79. Darland DC, Massingham LJ and Smith SR et al (2003) Pericyte production of cell-associated VEGF is differentiation-dependent and is associated with endothelial survival Dev Biol 264 275–88 DOI: 10.1016/j.ydbio.2003.08.015 PMID: 14623248
80. Yonenaga Y, Mori A and Onodera H et al (2005) Absence of smooth muscle actin-positive pericyte coverage of tumor vessels correlates with hematogenous metastasis and prognosis of colorectal cancer patients Oncol 69 159–66 DOI: 10.1159/000087840
81. Muller AJ and Scherle P (2006) Targeting the mechanisms of tumoral immune tolerance with small-molecules inhibitors Nature Rev 6 611–25
82. Le NT, Xue M and Castelnoble LA et al (2007) The dual personalities of matrix metalloproteinases in inflammation Front Biosci 12 1475–87 DOI: 10.2741/2161
83. Nelson D and Ganss R (2006) Tumor growth or regression: powered by inflammation J Leukoc Biol 80 685–690 DOI: 10.1189/jlb.1105646 PMID: 16864602
84. Lukanidin E and Sleeman JP (2012) Building the niche: the role of the S100 proteins in metastatic growth Sem Cancer Biol 22 216–25 DOI: 10.1016/j.semcancer.2012.02.006
85. Badolato R, Wang JM and Murphy WJ et al (1994) Serum amyloid A is a chemoattractant: induction of migration, adhesion, and tissue infiltration of monocytes and polymorphonuclear leukocytes J Exp Med 180 203–9 DOI: 10.1084/jem.180.1.203 PMID: 7516407 PMCID: 2191543
86. Malle E, Sodin-Semrl S and Kovacevic A (2009) Serum amyloid A: an acute-phase protein involved in tumour pathogenesis Cell Mol Life Sci 66 9–26 DOI: 10.1007/s00018-008-8321-x
87. Grivennikov SI and Karin M (2011) Inflammatory cytokines in cancer: tumour necrosis factor and interleukin 6 take the stage Ann Rheumat Dis 70 Suppl 1 104–8 DOI: 10.1136/ard.2010.140145
88. Yan HH, Pickup M and Pang Y et al (2010) Gr-1 CD11b myeloid cells tip the balance of immune protection to tumor promotion in the premetastatic lung Cancer Res 70 6139–49 DOI: 10.1158/0008-5472.CAN-10-0706 PMID: 20631080 PMCID: 4675145
89. Whiteside TL, Schuler P and Schilling B (2012) Induced and natural regulatory T cells in human cancer Expert Opin Biol Ther 12 1383–97 DOI: 10.1517/14712598.2012.707184 PMID: 22849383 PMCID: 3730844
90. Gasteiger G, Hemmers S and Firth MA et al (2013) IL-2-dependent tuning of NK cell sensitivity for target cells is controlled by regulatory T cells J Exp Med 210 1179–87 DOI: 10.1084/jem.20122571 PMID: 23650439 PMCID: 3674698
91. Bates GJ, Fox SB and Han C et al (2006) Quantification of regulatory T cells enables the identification of high-risk breast cancer patients and those at risk of late relapse J Clin Oncol 24 5373–80 DOI: 10.1200/JCO.2006.05.9584 PMID: 17135638
92. Frey DM, Droeser RA and Viehl CT et al (2010) High frequency of tumor-infiltrating FOXP3( ) regulatory T cells predicts improved survival in mismatch repair-proficient colorectal cancer patients Int J Cancer 126 2635–43
93. von Boehmer H and Daniel C (2013) Therapeutic opportunities for manipulating T (Reg) cells in autoimmunity and cancer Nature Rev Drug Discov 12 51–63 DOI: 10.1038/nrd3683
94. Hiratsuka S, Watanabe A and Sakura Y et al (2013) The S100A8 serum amyloid A3-TLR4 paracrine cascade establishes premetastatic phase Nat Cell Biol 10 1349–55 DOI: 10.1038/ncb1794
95. Hiratsuka S, Ishibashi S and Tomita T et al (2013) Primary tumours modulate innate immune signalling to create pre-metastatic vascular hyperpermeability foci Nat Commu 4 1853 DOI: 10.1038/ncomms2856
96. Hanahan D and Weinberg RA (2000) The hallmarks of cancer Cell 100 57–70 DOI: 10.1016/S0092-8674(00)81683-9 PMID: 10647931
97. Thiery JP and Sleeman JP (2006) Complex networks orchestrate epithelial-mesenchymal transitions Nat Rev Mol Cell Biol 7 131–42 DOI: 10.1038/nrm1835 PMID: 16493418
98. Hay ED (1995) An overview of epithelio-mesenchymal transformation Acta Anat (Basel) 154 8–20 DOI: 10.1159/000147748
99. Gabrilovich DI, Ostrand-Rosenberg S and Bronte V (2012) Coordinated regulation of myeloid cells by tumours Nat Rev Immunol 12 253–68 DOI: 10.1038/nri3175 PMID: 22437938 PMCID: 3587148
100. Condeelis J and Pollard JW (2006) Macrophages: obligate partners for tumor cell migration, invasion, and metastasis Cell 124 263–6 DOI: 10.1016/j.cell.2006.01.007 PMID: 16439202
101. Thiery JP, Acloque H and Huang RY et al (2009) Epithelial-mesenchymal transitions in development and disease Cell 139 871–90 DOI: 10.1016/j.cell.2009.11.007 PMID: 19945376
102. Bonde AK, Tischler V and Kumar S et al (2012) Intratumoral macrophages contribute to epithelial-mesenchymal transition in solid tumors BMC Cancer 12 35 DOI: 10.1186/1471-2407-12-35 PMID: 22273460 PMCID: 3314544
103. Labelle M, Begum S and Hynes RO (2011) Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis Cancer Cell 20 576–90 DOI: 10.1016/j.ccr.2011.09.009 PMID: 22094253 PMCID: 3487108
104. Gocheva V, Wang HW and Gadea BB et al (2010) IL-4 induces cathepsin protease activity in tumor-associated macrophages to promote cancer growth and invasion Genes Dev 24 241–55 DOI: 10.1101/gad.1874010 PMID: 20080943 PMCID: 2811826
105. Huber MA, Kraut N and Beug H (2005) Molecular requirements for epithelial-mesenchymal transition during tumor progression Curr Opin Cell Biol 17 548–58 DOI: 10.1016/j.ceb.2005.08.001 PMID: 16098727
106. Thiery JP (2002) Epithelial-mesenchymal transitions in tumour progression Nat Rev Cancer 2 442–54 DOI: 10.1038/nrc822 PMID: 12189386
107. Tsai JH and Yang J (2013) Epithelial-mesenchymal plasticity in carcinoma metastasis Genes Dev 27 2192–206 DOI: 10.1101/gad.225334.113 PMID: 24142872 PMCID: 3814640
108. Bullock MD, Sayan AE and Packham GK et al (2012) MicroRNAs: critical regulators of epithelial to mesenchymal (EMT) and mesenchymal to epithelial transition (MET) in cancer progression Biol Cell 104 3–12 DOI: 10.1111/boc.201100115
109. De Wever O, Pauwels P and De Craene B et al (2008) Molecular and pathological signatures of epithelial-mesenchymal transitions at the cancer invasion front Histochem Cell Biol 130 481–94 DOI: 10.1007/s00418-008-0464-1 PMID: 18648847 PMCID: 2522326
110. Kim K, Lu Z and Hay ED (2002) Direct evidence for a role of beta-catenin/LEF-1 signaling pathway in induction of EMT Cell Biol Int 26 463–76 DOI: 10.1006/cbir.2002.0901 PMID: 12095232
111. He TC, Sparks AB and Rago C et al (1998) Identification of c-MYC as a target of the APC pathway Science 281 1509–12 DOI: 10.1126/science.281.5382.1509 PMID: 9727977
112. Omary MB, Coulombe PA and McLean WH (2004) Intermediate filament proteins and their associated diseases N Engl J Med 351 2087–100 DOI: 10.1056/NEJMra040319 PMID: 15537907
113. Tania M, Khan MA and Fu J (2014) Epithelial to mesenchymal transition inducing transcription factors and metastatic cancer Tumour Biol 35 7335–42 DOI: 10.1007/s13277-014-2163-y PMID: 24880591
114. Kajita M, McClinic KN and Wade PA (2004) Aberrant expression of the transcription factors snail and slug alters the response to genotoxic stress Mol Cell Biol 24 7559–66 DOI: 10.1128/MCB.24.17.7559-7566.2004 PMID: 15314165 PMCID: 506998
115. Iwatsuki M, Mimori K and Yokobori T et al (2010) Epithelial-mesenchymal transition in cancer development and its clinical significance Cancer Sci 101 293–9 DOI: 10.1111/j.1349-7006.2009.01419.x
116. Batlle E, Sancho E and Francí C et al (2000) The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells Nat Cell Biol 2 84–9 DOI: 10.1038/35000034 PMID: 10655587
117. Cano A, Pérez-Moreno MA and Rodrigo I et al (2000) The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression Nat Cell Biol 2 76–83 DOI: 10.1038/35000025 PMID: 10655586
118. Eger A, Aigner K and Sonderegger S et al (2005) DeltaEF1 is a transcriptional repressor of E-cadherin and regulates epithelial plasticity in breast cancer cells Oncogene 24 2375–85 DOI: 10.1038/sj.onc.1208429 PMID: 15674322
119. Perez-Moreno MA, Locascio A and Rodrigo I et al (2001) A new role for E12/E47 in the repression of E-cadherin expression and epithelial-mesenchymal transitions J Biol Chem 276 27424–31 DOI: 10.1074/jbc.M100827200 PMID: 11309385
120. Comijn J, Berx G and Vermassen P et al (2001) The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion Mol Cell 7 1267–78 DOI: 10.1016/S1097-2765(01)00260-X PMID: 11430829
121. Yang J, Mani SA and Donaher JL et al (2004) RATwist, a master regulator of morphogenesis, plays an essential role in tumor metastasis Cell 117 927–39 DOI: 10.1016/j.cell.2004.06.006 PMID: 15210113
122. Oda H, Tsukita S and Takeichi M (1998) Dynamic behavior of the cadherin-based cell-cell adhesion system during Drosophila gastrulation Dev Biol 203 435–50 DOI: 10.1006/dbio.1998.9047 PMID: 9808792
123. Thiery JP and Sleeman JP (2006) Complex networks orchestrate epithelial-mesenchymal transitions Nat Rev Mol Cell Biol 7 131–42 DOI: 10.1038/nrm1835 PMID: 16493418
124. Cheng GZ, Chan J and Wang Q et al (2007) Twist transcriptionally up-regulates AKT2 in breast cancer cells leading to increased migration, invasion, and resistance to paclitaxel Cancer Res 67 1979–87 DOI: 10.1158/0008-5472.CAN-06-1479 PMID: 17332325
125. Park SM, Gaur AB and Lengyel E et al (2008) The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2 Genes Dev 22 894–907 DOI: 10.1101/gad.1640608 PMID: 18381893 PMCID: 2279201
126. Creighton CJ, Gibbons DL and Kurie JM (2013) The role of epithelial mesenchymal transition programming in invasion and metastasis: a clinical perspective Cancer Manag Res 5 187–95 DOI: 10.2147/CMAR.S35171
127. Ruan K, Bao S and Ouyang G (2009) The multifaceted role of periostin in tumorigenesis Cell Mol Life Sci 66 2219–30 DOI: 10.1007/s00018-009-0013-7 PMID: 19308325
128. Cichon MA, Nelson CM and Radisky DC (2015) Regulation of epithelial-mesenchymal transition in breast cancer cells by cell contact and adhesion Cancer Inform 9 14 (Suppl 3) 1–13
129. Liu ZJ, Semenza GL and Zhang HF (2015) Hypoxia-inducible factor 1 and breast cancer metastasis J Zhejiang Univ Sci B 16 32–43 DOI: 10.1631/jzus.B1400221 PMID: 25559953 PMCID: 4288942
130. Lim S, Becker A and Zimmar A et al (2013) SNAIL1 mediated epithelial mesenchymal transition confers chemoresistance PLos One 8 e66558 DOI: 10.1371/journal.pone.0066558
131. Li L, Bathia R (2011) Stem cell quiescence Clin Cancer Res 17 4936–41 DOI: 10.1158/1078-0432.CCR-10-1499 PMID: 21593194 PMCID: 3410675
132. Mallini P, Lennard T and Kirby J et al (2014) Epithelial-to-mesenchymal transition: what is the impact breast cáncer stem cell and drug resistance Cancer Treat Rev 40 341–8 DOI: 10.1016/j.ctrv.2013.09.008
133. Ribeiro AS and Paredes JP (2015) Cadherin linking breast cancer stem cells and invasion: a promising marker to identify an “intermediate/metastable” EMT state Front Oncol 4 371 DOI: 10.3389/fonc.2014.00371
134. Shirakihara T, Horiguchi K and Miyazawa K et al (2011) TGF-β regulates isoform switching of FGF receptors and epithelial-mesenchymal transition EMBO J 30(4) 783–95 DOI: 10.1038/emboj.2010.351 PMID: 21224849 PMCID: 3041949
135. Lee JK, Joo KM and Lee J et al (2014) Targeting the epithelial to mesenchymal transition in glioblastoma: the emerging role of MET signaling Onco Targets Ther 7 1933–44 PMID: 25364264 PMCID: 4211615
136. Jing Y, Han Z and Zhang S et al (2011) Epithelial-mesenchymal transition in tumor microenvironment Cell Biosci 1 29 DOI: 10.1186/2045-3701-1-29 PMID: 21880137 PMCID: 3179439
137. Katsuno Y, Lamouille S and Derynck R (2013) TGF-β signaling and epithelial-mesenchymal transition in cancer progression Curr Opin Oncol 25 76–84 DOI: 10.1097/CCO.0b013e32835b6371
138. Sahlgren C, Gustafsson MV and Jin S et al (2008) Notch signaling mediates hypoxia-induced tumor cell migration and invasion Proc Natl Acad Sci U S A 105 6392–7 DOI: 10.1073/pnas.0802047105 PMID: 18427106 PMCID: 2359811
139. Lindsey S and Langhans SA (2014) Crosstalk of oncogenic signaling pathways during epithelial-mesenchymal transition Front Oncol 4 358 DOI: 10.3389/fonc.2014.00358
140. Felibert P, Quintana J and Arvelo F (2009) Metaloproteasas Propiedades, funciones y sus usos como blancos terapéuticos en el tratamiento antineoplásico Acta Científica Venezolana 60 11–21
141. Gill SE and Parks WC (2008) Metalloproteinases and their inhibitors: regulators of wound healing Int J Biochem Cell Biol 40 1334–47 DOI: 10.1016/j.biocel.2007.10.024
142. Manicone AM and Mc Guire JK (2008) Matrix metalloproteinases as modulators of inflammation Semin Cell Dev Biol 19 34–41 DOI: 10.1016/j.semcdb.2007.07.003
143. Kessenbrock K, Plaks V and Werb Z (2010) Matrix metalloproteinases: regulators of the tumor microenvironment Cell 141 52–67 DOI: 10.1016/j.cell.2010.03.015 PMID: 20371345 PMCID: 2862057
144. Xiao LJ, Lin P and Lin F et al (2012) ADAM17 targets MMP-2 and MMP-9 via EGFR-MEK-ERK pathway activation to promote prostate cancer cell invasion Int J Oncol 40 1714–24
145. Visse R and Nagase H (20003) Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function and biochemistry Circ Res 92 827–39
146. Ito I, Fixman ED and Asai K et al (2009) Platelet-derived growth factor and transforming growth factor-beta modulate the expression of matrix metalloproteinases and migratory function of human airway smooth muscle cells Clin Exp Allergy 39 1370–80 DOI: 10.1111/j.1365-2222.2009.03293.x PMID: 19522858
147. Park CH and Chung JH (2011) Epidermal growth factor-induced matrix metalloproteinase-1 expression is negatively regulated by p38 MAPK in human skin fibroblasts J Dermatol Sci 64 134–41 DOI: 10.1016/j.jdermsci.2011.07.002 PMID: 21872446
148. Stetler-Stevenson WG (2008) The tumor microenvironment: regulation by MMP-independent effects of tissue inhibitor of metalloproteinases-2 Cancer Metastasis Rev 27 57–66 DOI: 10.1007/s10555-007-9105-8
149. Hagemann T, Robinson SC and Schulz M et al (2004) Enhanced invasiveness of breast cancer cell lines upon co-cultivation with macrophages is due to TNF-alpha dependent up-regulation of matrix metalloproteases Carcinogenesis 25 1543–9 DOI: 10.1093/carcin/bgh146 PMID: 15044327
150. Gialeli C, Theocharis AD and Karamanos NK (2011) Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting FEBS J 278 16–27 DOI: 10.1111/j.1742-4658.2010.07919.x
151. Van Damme J, Struyf S and Opdenakker G (2004) Chemokineprotease interaccions in cancer Semin Cancer Biol 14 201–8 DOI: 10.1016/j.semcancer.2003.10.007 PMID: 15246056
152. Bourboulia D and Stetler-Stevenson WG (2010) Matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs): positive and negative regulators in tumor cell adhesion Semin Cancer Biol 20 1618 DOI: 10.1016/j.semcancer.2010.05.002
153. Noël A, Jost M and Maquoi E (2008) Matrix metalloproteinases at cancer tumor-host interface Semin Cell Dev Biol 19 52–60 DOI: 10.1016/j.semcdb.2007.05.011
154. Hua H, Li M and Luo T et al (2011) Matrix metalloproteinases in tumorigenesis: an evolving paradigm Cell Mol Life Sci 68 3853–68 DOI: 10.1007/s00018-011-0763-x PMID: 21744247
155. Chabottaux V and Noel A (2007) Breast cancer progression: insights into multifaceted matrix metalloproteinases Clin Exp Metastasis 24 647–56 DOI: 10.1007/s10585-007-9113-7 PMID: 17968664
156. Decock J, Hendrickx W and Vanleeuw U et al (2008) Plasma MMP1 and MMP8 expression in breast cancer: protective role of MMP8 against lymph node metastasis BMC Cancer 8 77 DOI: 10.1186/1471-2407-8-77 PMID: 18366705 PMCID: 2278147
157. Gonzalez LO, Corte MD and Vazquez J et al (2008) Study of matrix metalloproteinases and their tissue inhibitors in ductal in situ carcinomas of the breast Histopathology 53 403–415 DOI: 10.1111/j.1365-2559.2008.03136.x PMID: 18983606
158. Rucci N, Sanită P and Angelucci A (2011) Expanding view of the role of matrix metalloproteases in metastatic growth Curr Mol Med 11 609–22 DOI: 10.2174/156652411797536705 PMID: 21707518
159. Hua H, Li M and Luo T et al (2011) Matrix metalloproteinases in tumorigenesis: an evolving paradigm Cell Mol Life Sci 68 3853–68 DOI: 10.1007/s00018-011-0763-x PMID: 21744247
160. Sounni NE, Paye A and Host L et al (2011) MT-MMPS as regulators of vessel stability associated with angiogenesis Front Pharmacol 2 111 DOI: 10.3389/fphar.2011.00111 PMID: 21687519 PMCID: 3108474
161. Lin HC, Chang JH and Jain S et al (2001) Matrilysin cleavage of corneal collagen type XVIII NC1 domain and generation of a 28-kDa fragment Invest Ophthalmol Vis Sci 42 2517–24 PMID: 11581192
162. Kessenbrock K, Plaks V and Werb Z (2010) Matrix metalloproteinases: regulators of the tumor microenvironment Cell 141 52–67 DOI: 10.1016/j.cell.2010.03.015 PMID: 20371345 PMCID: 2862057
163. Hanahan D and Weinberg RA (2011) Hallmarks of cancer: the next generation Cell 144 646–74 DOI: 10.1016/j.cell.2011.02.013 PMID: 21376230
164. Ferrara N (2005) The role of VEGF in the regulation of physiological and pathological angiogenesis EXS 94 209–31
165. Weis SM and Cheresh DA (2011) Tumor angiogenesis: molecular pathways and therapeutic targets Nature Med 17 1359–70 DOI: 10.1038/nm.2537 PMID: 22064426
166. Semenza GL (2013) Cancer-stromal cell interactions mediated by hypoxia-inducible factors promote angiogenesis, lymphangiogenesis, and metastasis Oncogene 32 4057–63 DOI: 10.1038/onc.2012.578
167. Cuiffo BG and Karnoub AE (2012) Mesenchymal stem cells in tumor development: emerging roles and concepts Cell Adh Migr 6 220–30 DOI: 10.4161/cam.20875 PMID: 22863739 PMCID: 3427236
168. Alitalo A and Detmar M (2012) Interaction of tumor cells and lymphatic vessels in cancer progression Oncogene 31 4499–508 DOI: 10.1038/onc.2011.602
169. Arvelo F, Cotte Carlos and Sojo Felipe (2014) Células Madre y Cáncer Invest Clín 55 371–91
170. Schoppmann SF, Birner P and Stöckl J et al (2002) Tumor-associated macrophages express lymphatic endothelial growth factors and are related to peritumoral lymphangiogenesis Am J Pathol 161 947–56 DOI: 10.1016/S0002-9440(10)64255-1 PMID: 12213723 PMCID: 1867252
171. Hanahan D and Folkman J (1996) Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis Cell 86 353–64 DOI: 10.1016/S0092-8674(00)80108-7 PMID: 8756718
172. Arvelo F (2009) Hipoxia en la malignidad del cáncer Invest Clin 50 529–46
173. Goodsell D (2003) The molecular perspective: VEGF and angiogenesis Stem Cells 21 118–9 DOI: 10.1634/stemcells.21-1-118 PMID: 12529559
174. North S, Moenner M and Bikfalvi A (2005) Recent developments in the regulation of the angiogenic switch by cellular stress factors in tumors Cancer Lett 218 1–14 DOI: 10.1016/j.canlet.2004.08.007 PMID: 15639335
175. Klein CA (2010) Framework models of tumor dormancy from patient-derived observations Curr Opin Genet Dev 21 42–49 DOI: 10.1016/j.gde.2010.10.011 PMID: 21145726
176. Kienast Y, von Baumgarten L and Fuhrmann M et al (2010) Real-time imaging reveals the single steps of brain metastasis formation Nature Med 16(1) 116–22 DOI: 10.1038/nm.2072
177. Schrader J, Gordon-Walker TT and Aucott RL et al (2011) Matrix stiffness modulates proliferation, chemotherapeutic response, and dormancy in hepatocellular carcinoma cells Hepatology 53 1192–205 DOI: 10.1002/hep.24108 PMID: 21442631 PMCID: 3076070
178. Barkan D, El Touny LH and Michalowski AM et al (2010) Metastatic growth from dormant cells induced by a col-I-enriched fibrotic environment Cancer Res 70 5706–16 DOI: 10.1158/0008-5472.CAN-09-2356 PMID: 20570886 PMCID: 3436125
179. Goetz JG, Minguet S and Navarro-Lerida I et al (2011) Biomechanical remodeling of the microenvironment by stromal caveolin-1 favors tumor invasion and metastasis Cell 146 148–63 DOI: 10.1016/j.cell.2011.05.040 PMID: 21729786 PMCID: 3244213
180. Fischer AN, Fuchs E and Mikula M et al (2007) PDGF essentially links TGF-beta signaling to nuclear beta-catenin accumulation in hepatocellular carcinoma progression Oncogene 26 3395–405 DOI: 10.1038/sj.onc.1210121
181. Barkan D, El Touny LH and Michalowski AM et al (2010) Metastatic growth from dormant cells induced by a col-I-enriched fibrotic environment Cancer Res 70 5706–16 DOI: 10.1158/0008-5472.CAN-09-2356 PMID: 20570886 PMCID: 3436125
182. Barkan D, Green JE and Chambers A F (2010) Extracellular matrix: a gatekeeper in the transition from dormancy to metastatic growth Eur J Cancer 46 1181–8 DOI: 10.1016/j.ejca.2010.02.027 PMID: 20304630 PMCID: 2856784
183. Mejlvang J, Kriajevska M and Vandewalle C et al (2007) Direct repression of cyclin D1 by SIP1 attenuates cell cycle progression in cells undergoing an epithelial mesenchymal transition Mol Biol Cell 18 4615–624 DOI: 10.1091/mbc.E07-05-0406 PMID: 17855508 PMCID: 2043563
184. Muller-Hermelink N, Braumuller H and Pichler B et al (2008) TNFR1 signaling and IFN-gamma signaling determine whether T cells induce tumor dormancy or promote multistage carcinogenesis Cancer Cell 13(6) 507–18 DOI: 10.1016/j.ccr.2008.04.001 PMID: 18538734
185. Tran DD, Corsa CA and Biswas H et al (2011) Temporal and spatial cooperation of Snail1 and Twist1 during epithelial–mesenchymal transition predicts for human breast cancer recurrence Mol Cancer Res 9 1644–57 DOI: 10.1158/1541-7786.MCR-11-0371 PMID: 22006115
186. Vega S, Morales AV and Ocana OH et al (2004) Snail blocks the cell cycle and confers resistance to cell death Genes Dev 18(10) 1131–43 DOI: 10.1101/gad.294104 PMID: 15155580 PMCID: 415638
187. Holmgren L, O’Reilly MS and Folkman J (1995) Dormancy of micrometastases: balanced proliferation and apoptosis in the presence of angiogenesis suppression Nature Med 1 149–53 DOI: 10.1038/nm0295-149 PMID: 7585012
188. Almog N, Henke V and Flores L et al (2006) Prolonged dormancy of human liposarcoma is associated with impaired tumor angiogenesis FASEB J 20 947–9 DOI: 10.1096/fj.05-3946fje PMID: 16638967
189. Dameron K, Volpert O and Tainsky M et al (1994) Control of angiogenesis in fibroblast by p53 regulation of trombospondin-1 Science 265 1582–4 DOI: 10.1126/science.7521539 PMID: 7521539
190. Weinstat-Saslow DL, Zabrenetzky VS and VanHoutte K et al (1994) Transfection of thrombospondin 1 complementary DNA into a human breast carcinoma cell line reduces primary tumor growth, metastatic potential, and angiogenesis Cancer Res 54 6504–11 PMID: 7527299
191. Ghajar CM, Peinado H and Mori H et al (2013) The perivascular niche regulates breast tumour dormancy Nature Cell Biol 15 807–17 DOI: 10.1038/ncb2767 PMID: 23728425 PMCID: 3826912
192. Indraccolo S, Minuzzo S and Masiero M et al (2009) Cross-talk between tumor and endothelial cells involving the Notch3-Dll4 interaction marks escape from tumor dormancy Cancer Res 69 1314–23 DOI: 10.1158/0008-5472.CAN-08-2791 PMID: 19208840
193. Lindau D, Gielen P and Kroesen M et al (2013) The immunosuppressive tumour network: myeloid-derived suppressor cells, regulatory T cells and natural killer T cells Immunol 138 105–15 DOI: 10.1111/imm.12036
194. Almand B, Clark JI and Nikitina E et al (2001) Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer J Immunol 166 678–89 DOI: 10.4049/jimmunol.166.1.678
195. Talmadge JE and Gabrilovich DI (2013) History of myeloid-derived suppressor cells Nature Rev Cancer 13 739–52 DOI: 10.1038/nrc3581
196. Gabrilovich DI, Ostrand-Rosenberg S and Bronte V (2012) Coordinated regulation of myeloid cells by tumours Nat Rev Immunol 12 253–68 DOI: 10.1038/nri3175 PMID: 22437938 PMCID: 3587148
197. Gabrilovich DI, Velders MP and Sotomayor EM et al (2005) Mechanism of immune dysfunction in cancer mediated by immature Gr-1 myeloid cells J Immunol 166 5398–406 DOI: 10.4049/jimmunol.166.9.5398
198. Sinha P, Clements VK and Ostrand-Rosenberg S (2005) Reduction of myeloid-derived suppressor cells and induction of M1 macrophages facilitate the rejection of established metastatic disease J Immunol 174 636–45 DOI: 10.4049/jimmunol.174.2.636 PMID: 15634881
199. Liu C, Yu S and Kappes J et al (2007) Expansion of spleen myeloid suppressor cells represses NK cell cytotoxicity in tumor bearing host Blood 109 4336–42 DOI: 10.1182/blood-2006-09-046201 PMID: 17244679 PMCID: 1885503
200. Diaz-Montero CM, Salem ML and Nishimura MI et al (2009) Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy Cancer Immunol Immunother 58 49–59 DOI: 10.1007/s00262-008-0523-4
201. Hwu P (2010) Treating cáncer by targeting the immune system N Engl J Med 363 779–81 DOI: 10.1056/NEJMe1006416 PMID: 20818880
202. Vonderheide RH and Glennie MJ (2013) Agonistic CD40 antibodies and cáncer therapy Clin Cancer Res 19 1035–43 DOI: 10.1158/1078-0432.CCR-12-2064 PMID: 23460534 PMCID: 3590838
203. McDermott J and Jimeno A (2015) Pembrolizumab: PD-1 inhibition as a therapeutic strategy in cancer Drugs Today (Barc) 51 7–20 DOI: 10.1358/dot.2015.51.1.2250387
204. Eustace AJ, Kennedy S and Larkin AM et al (2014) Predictive biomarkers for dasatinib treatment in melanoma Oncoscience 1 158–66 DOI: 10.18632/oncoscience.20
205. Król M, Majchrzak K and Mucha J et al (2013) CSF-1R as an inhibitor of apoptosis and promoter of proliferation, migration and invasion of canine mammary cancer cells BMC Vet Res 5(9) 65 DOI: 10.1186/1746-6148-9-65
206. Rashdan S and Hanna N (2014) Nintedanib for the treatment of non-small-cell lung cancer Expert Opin Pharmacother 15(5) 729–739 Epub 2014 Mar 5 DOI: 10.1517/14656566.2014.897695 PMID: 24597846
207. Krzeszinski JY, Wei W and Huynh H et al (2014) miR-34a blocks osteoporosis and bone metastasis by inhibiting osteoclastogenesis and Tgif2 Nature 28 512(7515) 431–5 Epub 2014 Jun 25 DOI: 10.1038/nature13375
208. Ji Q, Liu X and Han Z et al (2015) Resveratrol suppresses epithelial-to-mesenchymal transition in colorectal cancer through TGF-β1/Smads signaling pathway mediated Snail/E-cadherin expression BMC Cancer 15 97 DOI: 10.1186/s12885-015-1119-y
209. Sun XD, Liu XE and Huang DS (2013) Curcumin reverses the epithelial-mesenchymal transition of pancreatic cancer cells by inhibiting the Hedgehog signaling pathway Oncol Rep 29 2401–7 PMID: 23563640