Targeting BRAF V600E in metastatic colorectal cancer: where are we today?
Rodrigo Motta Guerrero1a, Veronica Arnao Labajos1b, Sophia Lozano Ballena2c, Carlos Aliaga Macha3d, Miguel Sotelo Lezama3e, Cristian Pacheco Roman1f, Paola Montenegro Beltran1g and Alejandro Figueroa Torrejon3h
1Instituto Nacional de Enfermedades Neoplásicas, Surquillo 15038, Peru
2Hospital Almanzor Aguinaga Asenjo, Chiclayo 14001, Peru
3Centro Oncológico ALIADA, San Isidro 15036, Peru
ahttps://orcid.org/0000-0002-8086-3513
bhttps://orcid.org/0000-0001-7079-1010
chttps://orcid.org/0000-0002-7868-6802
dhttps://orcid.org/0000-0003-0237-7058
ehttps://orcid.org/0000-0002-8861-9355
fhttps://orcid.org/0000-0003-2359-5126
ghttps://orcid.org/0000-0002-1484-9537
hhttps://orcid.org/0000-0001-8466-0097
Abstract
Colorectal cancer (CRC) is the second most frequent cause of direct cancer death worldwide. The study of the molecular state of oncogenes has predictive and prognostic value in metastatic CRC (mCRC). The B-raf proto-oncogene (BRAF) gene mutation represents the 8%–12% of all mutations in mCRC. The BRAF V600E mutation, considered the most common alteration of BRAF, corresponds to a constitutive kinase with a high activating capacity of the RAS/RAF/MEK/ERK pathway after a cascade of successive phosphorylations in the transcription of genes. BRAF V600E mutation is more prevalent in women, elderly, right-sided colon cancer and Caucasian population. Unfortunately, it is considered a poor predictive and prognosis biomarker. Patients with mCRC BRAF V600E mutated (BRAFm) are generally associated with poor response to chemotherapy and short progression-free survival and overall survival. Recently, randomised clinical trials have studied the combination of different chemotherapy regimens with angiogenic inhibitors in mCRC BRAFm. In addition, new anti-BRAF and immunotherapy agents have also been studied in this population, with positive results. The objective of this review is to acknowledge the biology and molecular pathway of BRAF, critically analyse the clinical trials and the therapy options published until today and evaluate the options of treatment according to the patient’s clinical presentation.
Keywords: colorectal neoplasms, antineoplastic agents, drug therapy, immunotherapy
Correspondence to: Rodrigo Motta Guerrero
Email: rodrigo.motta@aliada.com
Published: 15/12/2022
Received: 05/09/2022
Publication costs for this article were supported by ecancer (UK Charity number 1176307).
Copyright: © the authors; licensee ecancermedicalscience. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Introduction
Colorectal cancer (CRC) is one of the most frequent neoplasms diagnosed worldwide. GLOBOCAN reports the diagnosis of more than 1.9 million new CRC cases during the year 2020, representing 10% of all new cases of malignant neoplasms. In addition, CRC is the second leading cause of direct cancer death and 9.4% of all deaths from malignant neoplasms [1]. Currently, the molecular profile of all patients with metastatic CRC (mCRC) is evaluated in daily clinical practice. Mutation of the KRAS (40%), BRAF (8%–12%) and NRAS (5%–10%) oncogenes plays a role in carcinogenesis and has predictive and prognostic value. Microsatellite instability (MSI) is described as a hypermutability phenotype resulting from deficiency of the germline DNA mismatch repair (dMMR) system (3%–5%) and sporadic cases (10%–15%). MSI is considered a biomarker with predictive value of response to immunotherapy, which is why it is also being studied in mCRC. The study of the mutational status makes possible to select the best treatment option (chemotherapy, monoclonal antibodies, immunotherapy) for each patient in daily clinical practice [2–7]. The mutation value of other oncogenes such as HER-2 (2%), MET (2%), ROS-1 (0.2%–2.4%) and NTRK (0.2%–2.4%) is currently under evaluation [8].
Patients with mutated BRAF generally correspond to a population with poor prognosis. Transversion at residue 1799 (T1799A) leads to amino acid substitution from valine to glutamic acid at codon 600 (p. V600E) in exon 15 (BRAF V600E), resulting in a highly active constitutive kinase. BRAF V600E is considered the most frequent alteration, representing >90% of all BRAF mutations [9]. This mutation is more prevalent in patients aged above 60 years, females and Caucasians. BRAF V600E is associated with right-sided cancer, mucinous and poorly differentiated histology, saw-tooth architecture, high CpG island methylator phenotype (CIMP) levels and MSI-high (MSI-h). Patients with BRAF V600E mutation tend to metastasise more frequently to the peritoneum and distant lymph nodes. This biomarker is associated with short survival (approximately 10.4 months) [10]. BRAF non-V600E mutations (BRAF 594 and BRAF 596) are found in <2% of mCRC and still do not have a clearly defined prognostic and predictive value and constitute a subpopulation with different clinical-pathological characteristics [11]. Currently, there is great interest in studying the BRAF pathway, evaluating treatment with specific agents and developing new management approaches for this patient population.
BRAF gene and protein
The BRAF protooncogene is located in chromosome 7 (7q34) and encodes the cytoplasmic protein serine/threonine kinase. It is known that all RAF proteins (ARAF, BRAF and CRAF) can phosphorylate MEK (MEK1 and MEK2). BRAF is more commonly mutated and has the strongest activation capacity of MEK. BRAF is constituted by three domains: CR1, CR2 and CR3. CR1 encompasses the main RAS-binding domain, meanwhile CR2 corresponds to the serine/threonine-enriched regulatory domain. Both are located in the N-terminus of the protein. CR3 is a serine/threonine catalytic kinase domain that is located at the C-terminal and is regulated via phosphorylation [12–15].
The BRAF signalling pathway
A cascade of successive phosphorylations of the constituent protein molecules in the activation of the RAS/RAF/MEK/ERK (mitogen-activated protein kinase (MAPK)) pathway regulates different mechanisms like cell proliferation, differentiation, apoptosis and survival (Figure 1). First, BRAF binds RAS-GTP, releasing the autoinhibitory CR1–CR3 interaction and cease of kinase inhibition. CR2 works as a hinge for CR1 and CR3. CR3 is a double-lobed structure that contains the kinase domain and spans the range of 457–717 amino acids. The N-terminal lobe binds ATP, while the C-terminal lobe binds substrate proteins. The two lobes flank the kinase active cleft with the active residue D576 facing into the cleft. The N-terminal lobe also contains the P-loop that stabilises ATP by electrostatic binding with phosphate group. Hydrophobic interactions also stabilise the interaction with the nucleoside ATP. After conformational changes, KRAS recruits and binds to cytosolic BRAF, forming an active homo/heterodimer with another component of the RAF family proteins. This homo/heterodimer phosphorylates and activates MEK kinases (MEK1 and MEK2) and ERK stimulating transcription factors involved in cell proliferation, differentiation, motility, apoptosis (BCL-2 regulator) and survival (via the HIPPO pathway) [12, 15].
Mutated BRAF and carcinogenesis
The pathogenesis of CRC is a multistep mutational pathway consequence to the progressive accumulation of genetic and epigenetic alterations. Chromosomal instability and MSI are two molecular mechanisms that describe the evolution of histological alterations to carcinoma. The epigenetic hypermethylation and the inactivation of the gene MLH1 triggers the malignant development of CRC. Similarly, mutations in MMR genes (MSH2, MLH1, MSH6, PMS2 and PMS1) lead to the accumulation of mutations favouring malignant transformation. Mutation of the Wnt signalling is one of the initial alterations of the MSI pathway that leads to the formation of an early lesion. The following BRAF mutation and gene alterations, such as IGF2R, TGFbR2 and BAX mutation allow the evolution to a late lesion and finally to carcinoma. The positive association between BRAF V600E mutation with MSI-h/dMMR tumours is well documented, as well as the mutual exclusivity with KRAS mutation. Hypermethylation is one of the molecular features of BRAF V600E mutation which was observed more frequently in CRC with high CIMP (77%), compared to low or negative CIMP (18% and 0%, respectively). Methylation of the CpG island in the promoter lesion ends in the silencing of tumour suppressor genes resulting in carcinogenesis. Therefore, MSI-h/BRAF mutated CRC occurs as a consequence of methylation of MLH1. This is why a high incidence of CpG island methylation is observed in BRAF V600E mutated CRC, regardless of MSI status. BRAF mutation is reported in sporadic CRC with hypermethylated phenotype, but not in hereditary CRC such as Lynch syndrome [12, 16].
BRAF mutation and new molecular classifications of CRC
The CRC Subtype Consortium identified four molecular subtypes of CRC: CMS1 (MSI), CMS2 (canonical), CMS3 (metabolic) and CMS4 (mesenchymal). The incidence of BRAF mutation varies according to the molecular subtype. BRAF mutation is more frequently detected (45%) in the CMS1 cluster; meanwhile is less than 10% and 5% in the CMS3/CMS4 and CMS2 clusters, respectively. However, it has been reported that response to target therapy is heterogeneous in the molecular subgroups that harbours BRAF V600E mutation [12]. Two different molecular subtypes of BRAF V600E mutations have been distinguished: BM1 and BM2. BM2 corresponds to the 66% of all BRAF mutated CRC and is characterised by a dysregulation of events related to cycle checkpoints. BM2 group is enriched in metabolic processes, high levels of CDK1 and low levels of cyclin-D1. BM1 corresponds to the remaining 33% of BRAF mutations. BM1 is characterised by the activation of the KRAS/AKT pathway, alteration of mTOR/4EBP1 and enhancement of EMT. BM1 group shows a stronger immunological profile (activation of IL2/STAT5, IL6/JAK/STAT3 pathways, TNF-α signalling through NF-kB and allograft rejection), enrichment in angiogenesis and TGF-β-mediated processes. Although the BM1 subtype appears to have worse prognosis, MSI status remains the dominant prognostic factor [12, 16, 17]. BM1 and BM2 frequency differs between each molecular subtype. The majority of patients with BM1 and BM2 were classified as CMS1 (70%) and CMS4 (17%), meanwhile few were found in CMS3 (5%) and CMS2 (2%). Interestingly, all CMS4 BRAF mutations are classified as BM1, while CMS1 BRAF mutations are distributed across BM1 and BM2 [12, 17].
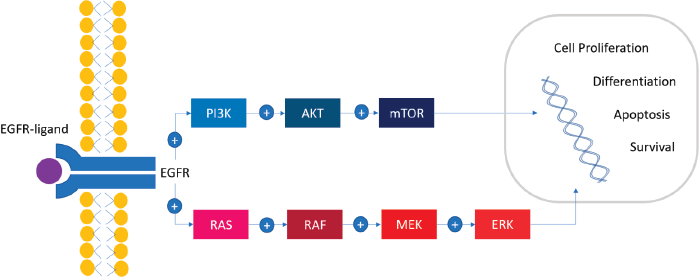
Figure 1. The BRAF signalling pathway. RAS/RAF/MEK/ERK signalling cascade, also known as the MAPK pathway, plays an important role in cellular proliferation, differentiation, survival and apoptosis. Multiple external sources (EGF, TGFα, epiregulin, etc.) bind to the external domain of EGFR with the consequent dimerisation of the receptor and the activation of the MAPK pathway. The activation of PI3K/AKT/mTOR pathway has been implicated as a mechanism of resistance.
BRAF mutations and microsatellite instability in CRC
Microsatellites are repeating DNA sequences of 1–6 nucleotides in the coding and non-coding regions of the genome. The DNA mismatch repair system is responsible for correcting errors in DNA replication. MSI-h is a hypermutability condition resulting from DNA mismatch repair system deficiency. It is associated with high number of tumoral neoantigens and lymphocytes T infiltration. In addition, MSI-h tumours have high angiogenic potential and high microvascular density that can facilitate a local inflammatory response and metastasis. MSI-h frequency in advance stages of CRC is approximately 15%. About 3%–5% of mCRC have MSI-h due to a germline mutation of the mismatch repair genes (Lynch syndrome) or subsequently to a somatic inactivation of a gene (10%–15%), most commonly through hypermethylation of the MLH1 promoter region. The somatic inactivation of mismatch repair genes is strongly associated with BRAF V600E mutation (60%), which is practically absent in Lynch syndrome. Patients with MSI-h and mutated BRAF share similar clinicopathological characteristics such as old age, female gender, right sidedness, mucinous histology, poor cell differentiation, high-grade intratumoral lymphocytes and peritumoral lymphoid reactions. The predictive and prognostic value of MSI-h and BRAF mutation is a topic of interest in the clinical practice [7, 18].
BRAF V600E mutated mCRC treatment strategies
Patients with mutated BRAF V600E mCRC tend to achieve median progression-free survival (mPFS) and overall survival (mOS) of only 4–6 and 10.4 months with conventional first-line therapies, respectively. The short survival presented in this population with poor prognosis led to the early use of aggressive therapy regimens [19]. For this reason, various treatment modalities have been evaluated in recent years. The main studies are summarised in Table 1.
Bevacizumab + chemotherapy: triplet versus duplet
The main problem in clinical trials is the small number of mutated BRAF patients included. An analysis of the FIRE-3 study evaluated the impact of KRAS and BRAF mutations on the efficacy of FOLFIRI-bevacizumab versus FOLFIRI-cetuximab combinations in mCRC. Forty-eight patients with mutated BRAF were included, of which only 25 patients received FOLFIRI-bevacizumab. This small group achieved an overall response rate (ORR) of 40%; while the mPFS and mOS were 6.6 and 13.7 months, respectively [20]. An analysis of the Australian phase III AGITG MAX study showed that BRAF mutation did not have predictive value for bevacizumab-chemotherapy combination. Mutated BRAF patients benefit similarly to wild-type BRAF patients with FOLFOX-bevacizumab regimen [21]. Other antiangiogenics (ramucirumab and aflibercept) have a mechanism of action similar to bevacizumab. Despite not having substantial evidence of their clinical benefit, they could potentially share similar efficacy in mutated BRAF population [22].
Retrospective evidence reported that the association of a chemotherapy triplet (FOLFOXIRI) with bevacizumab improved PFS in BRAF V600E mutated mCRC. A prospective phase II trial evaluated FOLFOXIRI-bevacizumab combination as first-line in 25 patients with mutated BRAF mCRC. The study was designed to detect an increase in the PFS rate, from 45% to 80%, after 6 months of treatment. The main objective was achieved and the PFS rate was 84% after 6 months of therapy. The median PFS and OS was 11.8 and 24.1 months, meanwhile ORR achieved was high (72%) and the disease control rate (DCR) was 88%. Although the efficacy results were superior to those obtained with FOLFOX-bevacizumab, FOLFIRINOX regimen was highly toxic. The most frequently reported grade 3–4 adverse events (AEs) were: neutropenia (40%), stomatitis (20%), diarrhoea (13%), asthenia (13%) and venous thrombosis (13%) [23]. Another prospective phase II trial evaluated FOLFOXIRI-bevacizumab in a group of 57 patients with mCRC, of which 10 had mutated BRAF tumours. After a median follow-up of 28.8 months, no benefit was reported in mPFS (12.8 versus 13.1 months, HR = 0.89, 95% CI: 0.41–1.91) and mOS (23.8 versus 90.9 months, HR = 0.76, 95% CI: 0.26–2.21) between mutated BRAF and wild-type BRAF patients. However, the response rate obtained in the mutated BRAF subpopulation was higher (90% versus 75%). Grade III–IV toxicity was similar to that previously recorded. Most of the patients had neutropenia (49%), diarrhoea (14%), hypertension (11%), deep vein thrombosis (7%), stomatitis (4%) and neurotoxicity (2%) [24].
Table 1. Most relevant clinical trials in mCRC with BRAF V600E mutation.
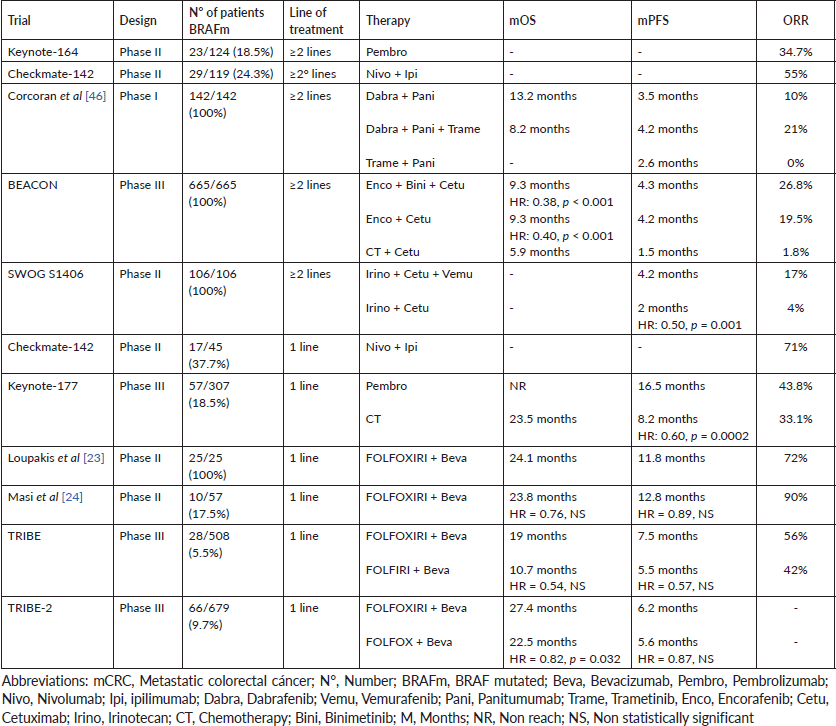
The TRIBE study was a prospective phase III trial that compared the efficacy of the combination FOLFOXIRI-bevacizumab versus FOLFIRI-bevacizumab in first-line. Of 508 included patients with mCRC, only 28 had mutated BRAF (12 received FOLFIRI-bevacizumab and 16 FOLFOXIRI-bevacizumab). FOLFOXIRI-bevacizumab had higher ORR (56% versus 42%; HR: 1.82, 95% CI: 0.38–8.78) and longer mPFS (7.5 versus 5.5 months; HR: 0.57, 95% CI: 0.27–1.23) and mOS (19 versus 10.7 months; HR: 0.54, 95% CI: 0.24–1.20). However, none of the differences were statistically significant, probably due to the limited number of mutated BRAF patients included [25]. Subsequently, the TRIBE-2 trial studied the FOLFOXIRI-bevacizumab combination versus FOLFOX-bevacizumab in first-line with 5FU-bevacizumab as maintenance therapy. At progression, FOLFOXIRI-bevacizumab reintroduction and FOLFIRI-bevacizumab sequence were indicated, respectively. The primary endpoint was PFS to second-line (PFS2). Six hundred and seventy-nine patients were enrolled and matched into the two treatment arms and the 10% (33) of patients in each arm had BRAF mutation (N = 66). There was no statistical difference between the median PFS2 (6.2 versus 5.6 months; HR = 0.87, 95% CI: 0.73–1.04, p = 0.11) between both regimens. FOLFOXIRI-bevacizumab achieved longer mPFS1 (12 versus 9.8 months; HR = 0.74, 95% CI: 0.63–0.86, p = 0.0002) and mOS (27.4 versus 22.5 months; HR = 0.82, 95% CI: 0.68–0.98, p = 0.032). In contrast to the TRIBE study, no benefit was found in mutated BRAF patients. It is suggested that a different comparator (FOLFOX in this study) or molecular heterogeneity of mutated BRAF tumours could explain these discordant results [26]. Zhou et al [27] performed a network meta-analysis to determine the most effective regimen in mCRC, including patients with mutated BRAF (N = 54). In this subpopulation, FOLFOXIRI-bevacizumab obtained longer mPFS than that obtained with bevacizumab duplets, although it did not reach statistical difference (HR: 0.64, 95% CI: 0.36–1.15). Recently, Cremolini et al [28] published a systematic review comparing the association of bevacizumab with duplet or triplet of chemotherapy in mCRC, including a considerable number of mutated BRAF patients (N = 115). FOLFOXIRI-bevacizumab did not improve mOS (13.5 versus 14.5 months; HR = 1.14, 95% CI: 0.75–1.73) compared to duplet-bevacizumab. No statistical difference was reported in PFS (HR = 0.84, 95% CI: 0.56–1.25) and ORR (HR = 1.42, 95% CI: 0.68–2.97) between both regimens. However, triplet-bevacizumab significantly increased grade 3–4 toxicity: neutropenia (45.8% versus 21.5%; p < 0.001), febrile neutropenia (6.3 versus 3.7%; p < 0.001), nausea (5.5% versus 3 %; p < 0.001), mucositis (5.1% versus 2.9%; p < 0.001) and diarrhoea (17.8% versus 8.4%; p < 0.001) [28]. Currently, the benefit of FOLFOXIRI-bevacizumab over duplets-bevacizumab regimens is not clear. The limited number of patients included in clinical trials limits conclusions.
Chemotherapy + EGFR inhibitors?
It is postulated that patients with mutated BRAF V600E mCRC are resistant to EGFR inhibition due to activation of the MAPK pathway secondary to the BRAF mutation. There is limited evidence on the clinical benefit of adding anti-EGFR therapy to chemotherapy treatment in BRAF-mutated mCRC. Pietrantonio [29] published a meta-analysis evaluating the addition of anti-EGFR agents to chemotherapy regimens in patients with mutated BRAF mCRC. Results showed no benefit in PFS (HR = 0.88, p = 0.33) or OS (HR = 0.91, p = 0.63) with EGFR inhibitors. This result reinforces the role of mutated BRAF as a predictor of resistance to anti-EGFR agents. However, Rowland et al [20] point out the lack of evidence to conclude whether EGFR inhibitors are effective in mutated BRAF tumours. An analysis of the FIRE-3 study compared FOLFIRI-cetuximab versus FOLFIRI-bevacizumab in mutated BRAF mCRC (N = 48) without finding significant differences. FOLFIRI-cetuximab and FOLFIRI-bevacizumab had similar median PFS (6.6 versus 6.6 months; HR = 0.84, 95% CI: 0.47–1.51, p = 0.56) and OS (12.3 versus 13.7 months; HR = 0.79, 95% CI: 0.43–1.46, p = 0.45). The German VOLFI study (N = 63) evaluated the benefit of FOLFOXIRI-panitumumab versus FOLFOXIRI in wild-type RAS mCRC. Only 16 patients with mutated BRAF were included in this trial. FOLFIRINOX-panitumumab obtained more than three times the ORR (71.4% versus 22.2%; OR: 8.75, p = 0.126) than the triplet without the anti-EGFR. A subsequent survival analysis determined that the study population achieved a higher mOS with panitumumab (35.7 versus 29.8 months; HR = 0.67, 95% CI: 0.41–1.11) in the cohort of patients with unresectable disease. Although there are conflicting results regarding the benefit of EGFR inhibitors, there is no robust evidence to support adding them to the combination of first-line cytotoxic agents for mutated BRAF mCRC [20, 29–31].
Immunotherapy: new alternative in MSI-h/dMMR
Immunotherapy has been evaluated as an alternative treatment in mCRC in clinical trials that included patients with BRAF mutation. The Keynote-164 trial (N = 124) studied pembrolizumab, anti-PD1 agent, in previously treated MSI-h/dMMR mCRC, including only 23 with mutated BRAF. Although the small sample of mutated BRAF patients, the results obtained in this subpopulation were interesting. The 55% (5 patients) of cohort A (≥2 prior lines of treatment) and 20% (1 patient) of cohort B (≥1 prior line of treatment) with BRAF mutation achieved response with pembrolizumab. In addition, pembrolizumab was safe in the population of therapy. Only 16% of patients had grade ≥ 3 AEs: pancreatitis (3%), fatigue (3%), hepatitis (2%), pneumonitis (2%), asthenia (2%), arthralgia (2%) and skin toxicity (2%). Only the 3% of patients discontinued treatment due to toxicity [32]. The multicohort Checkmate-142 trial (N = 74) evaluated nivolumab, another anti-PD1 agent, in heavily pretreated MSI-high/dMMR mCRC. Only 16% (12) of patients had mutated BRAF. The 25% (3 patients) achieved ORR, while 75% (9 patients) achieved disease control beyond 12 weeks of therapy. Nivolumab was also well tolerated; the most frequent grade ≥ 3 AEs were elevated lipases (8%) and amylases (3%) [33]. Another of the cohorts of the Checkmate-142 trial (N = 119) studied the safety and efficacy of nivolumab associated with an anti-CTLA4 agent (ipilimumab). This cohort included 29 heavily pretreated patients with MSI-h/dMMR and mutated BRAF mCRC. In the mutated BRAF subpopulation; ORR and DCR were 55% and 79%, respectively. Nivolumab-ipilimumab was relatively more toxic than anti-PD1 monotherapy. The 20% of patients had grade ≥ 3 AEs: elevated AST (8%), elevated ALT (7%), diarrhoea (2%), fatigue (2%), pruritus (2%) and skin rash (2%). 13% of patients discontinued therapy due to toxicity [34]. Results from another Checkmate-142 cohort, presented at ASCO GI (2020), evaluated the efficacy of first-line nivolumab-ipilimumab. Of the 45 patients diagnosed with MSI-h/dMMR mCRC, only 17 patients had BRAF mutation. The ORR (77%) was the highest obtained to date with any treatment in mutated BRAF patients; however, the small number of patients included limits the knowledge of the real clinical benefit of this regimen. Apparently, the anti-PD1/anti-CTLA4 combination presented less toxicity than that reported in second line. The 16% had grade ≥ 3 AEs and only 7% discontinued treatment [35].
Immunotherapy was finally studied in a phase III trial in first-line. The Keynote-177 study compared pembrolizumab versus chemotherapy as first-line in MSI-h/dMMR mCRC. The pembrolizumab group achieved significantly longer mPFS (16.5 versus 8.2 months; HR: 0.60, 95% CI: 0.45–0.80; p = 0.0002) and ORR (43.8% versus 33.1%) than the patients who received chemotherapy. At the moment the results were published, the data for the survival analysis was still immature and median OS was not reached. Despite this, the study Keynote-177 meant a change in the treatment paradigm of MSI-h/dMMR mCRC. The 22% (34) of patients in the pembrolizumab group and 23% (34) in the chemotherapy group had mutated BRAF. It is remarking that benefit of PFS was reported both, in wild-type BRAF (HR: 0.50, 95% CI: 0.31–0.80) and in mutated BRAF (HR: 0.48, 95% CI: 0.27–0.86) patients. Though the main objective of the study was not to evaluate the efficacy of pembrolizumab in patients with mutated oncogenes, it is interesting to notice clinical benefit in this subpopulation. As described in the study Keynote-164, toxicity was low and manageable. Only 9% reported grade ≥3 AEs [36]. The last results updated were presented in the ‘Gastrointestinal Cancers Symposium’ of the year 2021. Patients who received pembrolizumab presented longer PFS with the second line of therapy (PFS2). In addition, the mOS was significantly longer with the anti-PD1 agent. The chemotherapy group reached mOS of 23.5 months, while the pembrolizumab group did not reach median OS (HR: 0.63, 95% CI: 0.45–0.88) yet [37]. A recent meta-analysis evaluated the prognostic and predictive value of the BRAF mutation in MSI-h/dMMR mCRC. Mutated BRAF patients had shorter survival than non-mutated BRAF patients in all clinical stages (I–IV) (HR: 1.57; 95% CI: 1.23–1.99). The ORR obtained was similar among mutated BRAF and wild-type BRAF patients treated with immunotherapy (OR: 1.04, 95% CI: 0.48–2.25). According to these results, survival of MSI-h/dMMR mCRC with BRAF mutation may significantly benefit with immunotherapy [38]. During the preparation of this review, no meta-analyses evaluating the efficacy of immunotherapy on PFS or OS in mutated BRAF/MSI-h/dMMR mCRC were found. The limited number of mutated BRAF patients included in clinical trials does not allow the development of more complex studies. However, the results suggest a potentially robust benefit with immunotherapy in this population.
BRAF inhibition: looking for the optimal combination
The efficacy of treatment with BRAF inhibitors was studied in small clinical trials, obtaining modest benefit in mutated BRAF V600E mCRC. A phase II trial (N = 10) reported that vemurafenib achieved mPFS of 2.1 and mOS of 7.7 months, without improving ORR. Another phase II trial (N = 21) reported similar results in survival (mPFS = 4.5 months and mOS = 9.3 months) and low ORR (5%) with vemurafenib [39, 40]. A phase I trial (N = 18) evaluated the efficacy of encorafenib, selective RAF kinase inhibitor, without positive results. Encorafenib achieved mPFS of 4 months, without improving ORR [41]. Preclinical studies suggest that BRAF inhibitors do not produce sustained inhibition of the MAPK pathway, which leads to a lack of response to these agents [42].
Laboratory studies suggest that the combined BRAF and MEK inhibition could achieve greater suppression of the MAPK pathway and greater antitumour efficacy. Unfortunately, the results were not the most favourable. The combination of BRAF inhibitors (dabrafenib) with MEK inhibitors (trametinib) was studied in 43 patients with heavily previously treated mutated BRAF mCRC (51% received ≥ 3 lines of therapy). Tumour samples were evaluated, showing lower levels of phosphorylated ERK compared to samples before the beginning of therapy (47%–24%). The mPFS was 3.5 months and the median duration of therapy was 3.6 months, meanwhile 23% (10 patients) continued therapy for more than 6 months. The 12% (5 patients) had partial response and 1 patient achieved complete response. Interestingly, the complete responder and two partial responders were reported to have PI3K pathway mutations. However, severe toxicity was significant with the combination. The 98% of patients presented AEs and 58% had grade ≥ 3 toxicity; the most frequent were: anaemia (16%), pyrexia (12%), vomiting (7%) and fatigue (7%). Pyrexia was the most common reason for discontinuing therapy (30%) [42].
Activation of the PI3K/AKT pathway has been identified as a mechanism of resistance to BRAF inhibitors in mutated BRAF mCRC. A recent publication reported that genetic alterations in EGFR and PI3K were associated with poor response to targeted treatment and the development of secondary resistance mutations. A phase Ib trial evaluated the safety of the encorafenib-cetuximab regimen (N = 26) compared with the same regimen associated with a PI3K inhibitor (alpelisib) (N = 28) in refractory mutated BRAF mCRC. The regimen including the PI3K inhibitor was well tolerated and only two patients reported dose-limiting toxicity. The combination with alpelisib achieved ORR of 18% and mPFS of 4.2 months. However, no further published results on PI3K inhibitors in mutated BRAF mCRC were found until the preparation of this review [43, 44].
Subsequently, preclinical studies demonstrated a decreased sensitivity to BRAF inhibitors with transient suppression of phosphorylated ERK, followed by the reactivation of RAS and C-RAF, mediated by the EGFR pathway. The combination of BRAF and EGFR inhibitors had a synergistic effect in vitro, resulting in sustained suppression of the MAPK pathway and the improvement of the efficacy of tumour growth inhibition [45]. These results led to the study of the addition of an EGFR inhibitor to the BRAF inhibition in clinical trials. A phase II trial (n = 27) evaluated the vemurafenib-cetuximab combination in pretreated mutated BRAF mCRC. Only one patient achieved partial response, but approximately half of the population had tumour shrinkage without achieving partial response. The median PFS and OS were 3.7 months (95% CI: 1.8–5.1) and 7.1 months (95% CI: 4.4–not reached), respectively [41]. Another trial evaluated the efficacy of BRAF inhibition, via anti-BRAF (dabrafenib) with anti-MEK (trametinib) agents in association with EGFR inhibition (panitumumab). One hundred and forty-two patients with previously treated BRAF V600E mutated mCRC (35% received ≥ 2 lines) were enrolled and distributed into three arms: dabrafenib-panitumumab, dabrafenib-panitumumab-trametinib and trametinib-panitumumab. The primary objective was to assess the ORR of the three combinations and dabrafenib-panitumumab-trametinib achieved the highest ORR (21%). Dabrafenib-panitumumab and trametinib-panitumumab had 10% and 0% of ORR, respectively. However, there was no superiority in PFS between the three arms. The mPFS was 4.2, 3.5 and 2.6 months with dabrafenib-panitumumab-trametinib, dabrafenib-panitumumab and trametinib-panitumumab combinations; respectively. OS data was still immature when the study was published; however, mOS was achieved in dabrafenib-panitumumab (13.2 months; 95% CI: 6.7–22 months) and trametinib-panitumumab (8.2 months; 95% CI: 6.7–22 months) groups. Dabrafenib-panitumumab-trametinib combination was associated with higher MAPK suppression (60%) compared to the dabrafenib-panitumumab (23%) and trametinib-panitumumab (41%) regimens. The addition of an EGFR inhibitor to BRAF inhibition seems to have a slight benefit in BRAF V600E mutated mCRC. However, the immature data do not allow establishing a clear clinical benefit in survival. We need a longer follow-up time to validate these results. The toxicity of the triplet was notoriously high, 70% presented AEs grade ≥ 3, the most frequent being: skin rash (11%), acneiform dermatitis (10%), fatigue (7%) and diarrhoea (7%) [46].
The phase III BEACON study evaluated the BRAF inhibition through the combination of a BRAF inhibitor (encorafenib) with a MEK inhibitor (binimetinib), and the inhibition of EGFR pathway (cetuximab) in previously treated with 1–2 lines mutated BRAF V600E mCRC. Six hundred and sixty-five patients were enrolled and randomised into three groups: the triplet group (encorafenib-binimetinib-cetuximab), the duplet group (encorafenib-cetuximab) and the control group (treatment of choice chosen by the investigator: cetuximab-irinotecan or cetuximab- FOLFIRI). The primary endpoints of the study were OS and ORR in the triplet group. The mOS (9 versus 5.4 months; HR: 0.52; 95% CI: 0.39–0.70; p < 0.001) and ORR (26% versus 2%; p < 0.001) were longer with encorafenib-binimetinib-cetuximab. The study was not designed to compare the outcomes between the triplet and duplet groups; but the OS and ORR obtained with triplet was similar to the result of encorafenib-cetuximab combination (ORR = 20%, mOS = 8.4 months). The mPFS was significantly higher in the triplet group (4.3 versus 1.5 months; HR: 0.38, 95% CI: 0.29–0.49, p < 0.001) and the doublet group (4.2 versus 1.5 months; HR: 0.4, 95% CI: 0.31–0.52, p < 0.001) compared to the control group. Regarding toxicity, 58% of patients in the triplet group, 50% in the duplet group and 61% in the control group presented grade 3–4 AEs. The most frequently reported AEs were: anaemia (11%), diarrhoea (10%), abdominal pain (6%), nausea (5%) and altered creatinine (5%). A subsequent safety analysis of the encorafenib-binimetinib-cetuximab regimen reported that five patients had dose-limiting toxicity; of which two presented retinopathies, one decreased left ventricular ejection fraction, and two infusional reactions related to cetuximab. The most commonly reported grade 3–4 AEs with the triplet were: fatigue (13%), anaemia (10%), increased creatinine phosphokinase (10%), increased AST (10%) and urinary tract infections (10%) [47]. The update of the study published in 2021 determined that the mOS was 9.3 months for the triplet group and 5.9 months for the control group (HR: 0.60, 95% CI: 0.47–0.75); meanwhile, the mOS for the duplet group was 9.3 months, also higher than the control group (HR: 0.61, 95% CI: 0.48–0.77). The ORR was 26.8% (95% CI: 21.1%–33.1%) for the triplet, 19.5% (95% CI: 14.5%–25.4%) for the duplet and 1.8% (95% CI: 0.5%–4.6%) for the control. The encorafenib-cetuximab combination improved OS, PFS and ORR similarly to the triplet, but with less toxicity, becoming the new standard of care in previously treated mutated BRAF V600E mCRC [48]. Subsequently, the ANCHOR study, a phase II trial, evaluated the same regimen in first-line for mutated BRAF V600E mCRC. The results of the first stage of this trial were presented at the ‘ESMO Gastrointestinal Congress’, reporting the results of 40 patients, who achieved ORR of 50% (95% CI: 33.8–66.2), with tumour reduction of 85%. However, the mPFS was only 4.9 months (95% CI: 4.4–8.1). Final results are still being waited today [49].
Recently, the combination of BRAF and EGFR inhibition in combination with chemotherapy has been studied. The SWOG-S1406 trial evaluated irinotecan-cetuximab versus irinotecan-cetuximab-vemurafenib treatment in 106 patients with mutated BRAF V600E mCRC, previously treated with 1–2 lines. Vemurafenib regimen achieved longer mPFS (4.2 versus 2 months; HR = 0.50, 95% CI: 0.32–0.76, p = 0.001) and higher ORR (17% versus 4%, p = 0.05) compared with irinotecan-cetuximab. A decreased frequency of circulating tumour BRAF V600E DNA was reported with vemurafenib (87% versus 0%, p < 0.001), with a poor incidence of acquired RAS alterations at progression. Grade 3–4 toxicity was more common in patients receiving vemurafenib; the most common were: neutropenia (30%), nausea (19%) and anaemia (13%). 22% of patients discontinued therapy due to toxicity [50].
What about non-V600E BRAF mutated mCRC treatment?
As previously described, a small percentage (<2%) of patients harbours BRAF non-V600E mutations. Mutated BRAF non-V600E tumours are associated with younger age, lower degree of cellular differentiation, less frequency mucinous histology, microsatellite stability, left-sided disease and a less aggressive evolution of cancer than patients with mutated BRAF V600E or wild-type BRAF tumours. BRAF non-V600E mutations are usually associated with significantly longer survival. However, because of the low frequency of these mutations, there is no randomised prospective data about potential treatment agents in this population. Recently, studying reports suggest that BRAF non-V600E tumours may be sensitive to EGFR inhibitors. However, the small sample size does not allow to make definitive conclusions. In addition, it has been described long-term response with regorafenib in one heavily pretreated mCRC patient with BRAF non-V600E. This result was attributed to the regorafenib’s efficacy targeting the epithelial to mesenchymal pathway in vitro. Further prospective studies are needed to find potential agents for these mutations [9, 11, 51–53].
Treatment in clinical practice
Patients with mutated BRAF mCRC correspond to a small population that has not been significantly represented in clinical trials of mCRC and has not been widely studied. Retrospective and prospective studies have reported a slight benefit in survival with cytotoxic treatment associated with bevacizumab in this poor prognosis population. The guide of the ‘Spanish Society of Medical Oncology’ (SEOM) and the ‘Consensus of Pan-Asian guidelines adapted by ESMO for the Management of metastatic colorectal cancer: JSMO-ESMO initiative endorsed by CSCO, KACO, MOS, SSO, and TOS’, both published in 2018, suggest the use of intense chemotherapy regimens (FOLFOXIRI) associated with bevacizumab in first-line therapy for BRAF mutated mCRC with good clinical status (ECOG 0–1), regardless of sidedness (right colon and left colon) (level of evidence IIB). The chemotherapy duplet associated with bevacizumab is suggested as the second option and the chemotherapy duplet without the anti-angiogenic agent as the third option. However, the ‘German evidence-based guideline for colorectal cancer’ (2019) acknowledges the positive results of the TRIBE study, but highlights that the small number of BRAF mutated patients included only allows for establishing treatment hypotheses [54–56]. The German guideline suggests that this population should receive the most effective treatment, such as the triplet, or be included in clinical trials (grade of recommendation B). The Japanese Society for Colorectal Cancer (JSCRC) (2019) guideline recommends the combination FOLFOXIRI-bevacizumab in the first-line treatment of BRAF mutated mCRC, according to the results of the TRIBE study [57]. The most recently published evidence, version 1 of the ‘National Comprehensive Cancer Network’ (NCCN) guideline (2022), recommends the FOLFOX or FOLFOXIRI regimens associated or not with an anti-angiogenic agent in patients who are candidates for receiving intense therapy (category II-A) [57]. This indication is more in line with the results of the TRIBE-2 study and the recently published meta-analyses by Zhou et al [27] and Cremolini et al [28]. Treatment with the FOLFOXIRI-bevacizumab regimen causes significant toxicity in daily clinical practice. Considering the latest publications of clinical trials and meta-analyses, it is consistent to consider the use of FOLFOXIRI-bevacizumab in selected patients (young, ECOG 0–1, no severe comorbidities) when the goal of treatment is to achieve response rate (symptomatic patients and/or extensive tumour involvement). In all other cases, the most appropriate indication is the use of duplet regimens with an angiogenic inhibitor. The combination of single chemotherapy agent with an angiogenic inhibitor is an alternative of treatment according to the patient’s clinical and molecular characteristics [58].
In daily practice, the mutational state of biomarkers (KRAS, NRAS, BRAF, MSI) of patients with mCRC is studied. Based on the results of the Keynote-177 study and the Checkmate-164 study, pembrolizumab and the nivolumab-ipilimumab combination are first-line treatment options for mCRC-high MSI/dMMR. Pembrolizumab was approved by the ‘Food and Drugs Administration’ (FDA) and by the European Medicines Agency (EMA), in June and December of the year 2020; respectively. The British guide ‘National Institute for Health and Care Excellence’ (NICE) (2021) recommends the use of pembrolizumab in untreated MSI-h/dMMR mCRC [59]. Similarly, the NCCN guideline (2022) suggests the use of pembrolizumab (preferred option) and nivolumab-ipilimumab as preferred first-line therapy in MSI-h/dMMR mCRC (both category II-A). FDA approved pembrolizumab and the combination nivolumab-ipilimumab for previously treated MSI-h/dMMR mCRC in May 2017 and July 2018, respectively [60]. The NCCN guideline (2022) recommends pembrolizumab (preferred) and nivolumab-ipilimumab as therapy options beyond the first line of treatment in MSI-h/dMMR mCRC (both with category II-A) [58]. Considering the design of the Keynote-177 trial (phase III with comparator) and the lower toxic profile with a single agent, the use of pembrolizumab as the agent of choice in this subpopulation of patients is valid. BRAF mutated patients without MSI-h/dMMR should receive combinations of duplet or triplet chemotherapy, ideally associated with an anti-angiogenic agent.
Recently, clinical trials have reported positive results with the use of regimens based on combinations of different agents. The FDA and EMA approved the encorafenib-cetuximab combination for the treatment of previously treated mCRC with BRAF V600E mutation, in April and June 2020, respectively. Both regulatory agencies approved the regimen based on the reported findings of the BEACON study [61, 62]. The British guide ‘National Institute for Health and Care Excellence’ (NICE) (2021) recommended the encorafenib-cetuximab combination for the treatment of adult patients with mutated BRAF mCRC who previously received systemic therapy. The suggested recommendation is based on the fact that the benefit provided in this population with a poor prognosis has been minimal in decades of mCRC studies and the encorafenib-cetuximab regimen is a paradigm shift in treatment. They also point out that mCRC and its treatment with conventional regimens affect the quality of life of patients. They emphasise that the toxicity of the encorafenib-cetuximab combination is manageable, which significantly improved the quality of life of the patients [63]. Similarly, the ‘National Comprehensive Cancer Network’ (NCCN) guideline (2022) recommends the combination of encorafenib (300 mg daily, PO) associated with cetuximab (400 mg/m2 IV, followed by 250 mg/m2 weekly) or panitumumab (6 mg/kg IV, every 14 days) as treatment options in patients with mutated BRAF V600E mCRC who progressed within 12 months of completing adjuvant chemotherapy or progressed to first-line oxaliplatin-based with/without irinotecan and with/without bevacizumab, or progressed to a subsequent irinotecan-based line (level of Evidence IIA). The encorafenib-cetuximab combination obtained interesting results in the BEACON study and is the regimen of choice after progression to first-line systemic therapy. The use of encorafenib-cetuximab could be considered when the patient progresses to adjuvant therapy with an oxaliplatin-associated fluoropyrimidine (FOLFOX/CAPOX) or relapses within 12 months of completing adjuvant FOLFOX or CAPOX, as option to treatment with FOLFIRI. Beyond the progression to encorafenib-cetuximab, a subsequent new line of therapy could be considered, according to ECOG and sequelae due to treatment toxicity. Alternatives include regorafenib or reintroduction of fluoropyrimidine therapy with or without oxaliplatin, associated or not with an anti-angiogenic agent (ramucirumab, aflibercept, reintroduction of bevacizumab) [58]. Summary in Figure 2.
Future perspectives
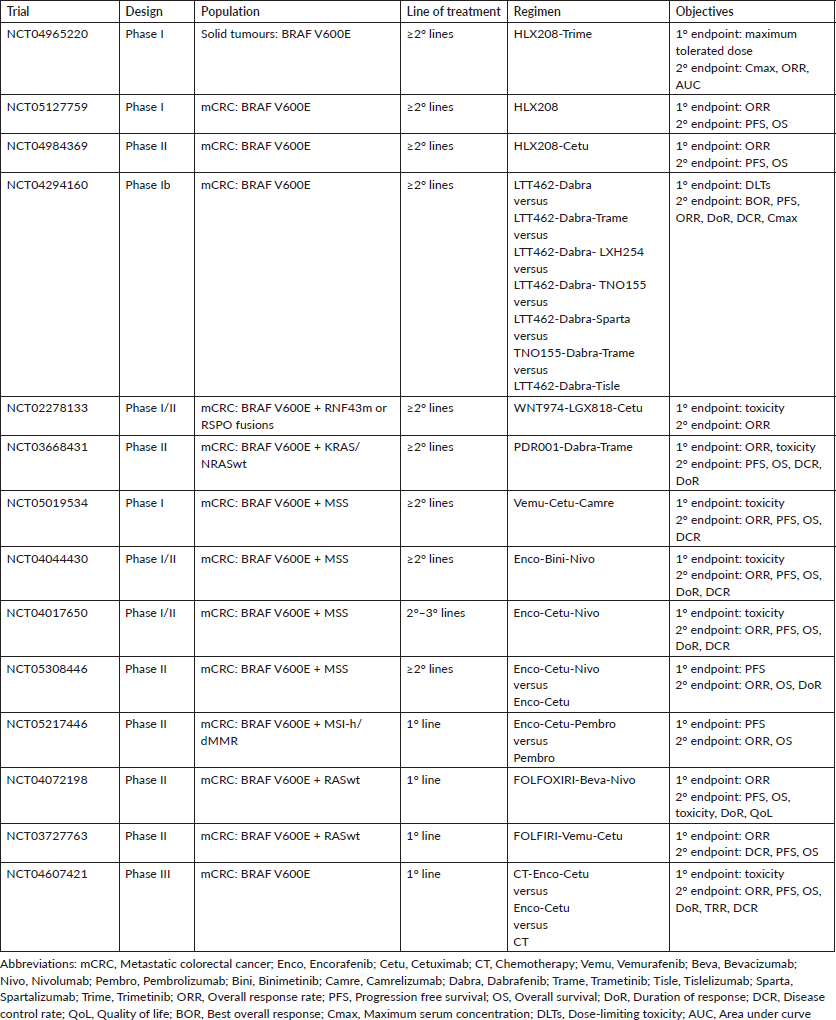
Studies that associate immunotherapy agents with BRAF inhibitors are currently being carried out. As previously described, MSI is associated with BRAF mutation, secondary to epigenetic inactivation of the mismatch repair protein MSH1, for which patients are reclassified in the molecular subgroup MCS1, characterised by high hypermethylation and response to immunotherapy. The SEAMARK trial (NCT05217446) is currently evaluating the combination encorafenib-cetuximab-pembrolizumab versus pembrolizumab alone in mCRC with IMS-h/dMMR. However, special interest is being added to the treatment of patients with microsatellite stable (MSS). Several clinical trials are evaluating the association of BRAF and EGFR inhibition with immunotherapy in heavily pretreated mCRC patients with MSS (NCT05019534, NCT05308446, NCT04017650). In addition, the NCT04044430 study is a phase I/II trial that evaluates the safety of the combination encorafenib-binimetinib associated with nivolumab in mutated BRAF V600E mCRC and MSS. The NIVACOR trial (NCT04072198), single-arm phase II study, in which the treatment with FOLFOXIRI-bevacizumab-nivolumab is being evaluated in mutated BRAF V600E patients. The study is still in the recruitment phase. A preliminary safety analysis of the first ten patients included reported a median of 5.5 cycles of treatment [17, 64, 65].
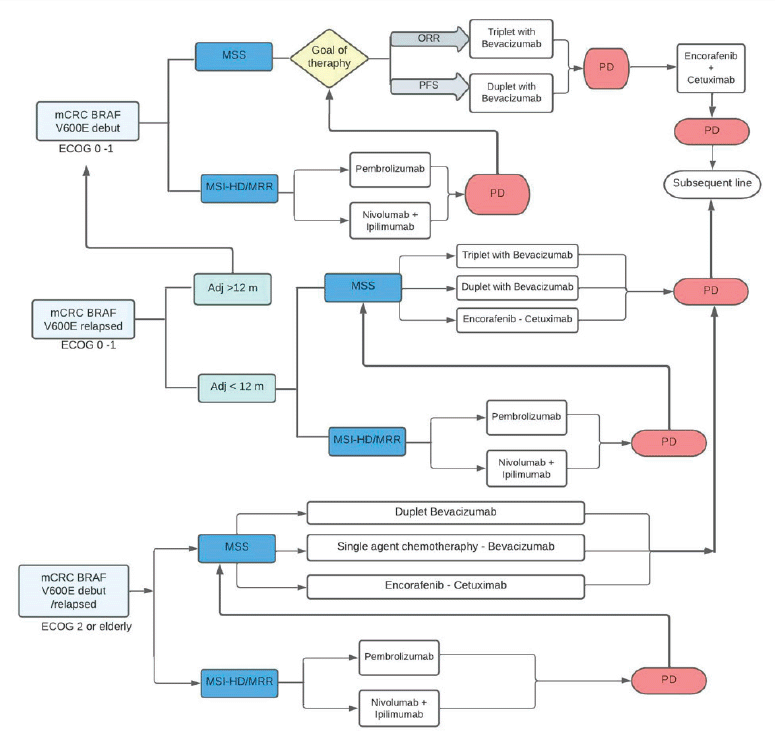
Figure 2. Algorithm of treatment in mutated BRAF V600E mCRC. mCRC, Metastatic colorectal cáncer; MSI-h, High microsatellite instability; dMMR, DNA mismatch repair; MSS, Microsatellite stable; ORR, Overall response rate; PFS, Progression free survival; PD, Progression of disease.
Other new therapy strategies are the new anti-BRAF agents, such as HLX208 alone or in combination with MEK or EGFR inhibitors (NCT04965220, NCT05127759, NCT04984369). Similarly; the safety and efficacy of ERK, RAF and SHP2 inhibitors are being evaluated in phase I clinical trial (NCT04294160). Looking at preclinical studies, ‘Wee1 and ERK1/2’ are points downstream of the BRAF pathway in the MAP kinase signalling cascade and are potentially important therapeutic targets, and based on this, AZD1775 (Wee1 inhibitor) and LY3214996 (ERK1/2 inhibitor) have been tested in phase I trials. In addition, the activation of the Wnt pathway by the RNF43 mutation or RSPO fusions may contribute to resistance in mutated BRAF mCRC. Under this premise, a phase I/II trial of combined therapy of Wnt inhibitor, WNT974 (porcupine inhibitor), with BRAF inhibition is being carried out (NCT02278133) [66, 67].
Based on the positive results obtained with SWOG 1406 trial, simultaneous EGFR and BRAF inhibition associated with cytotoxic agents is effective and safe in patients with mutated BRAF mCRC. These results put on hold the BREAKWATER study, a phase III trial that evaluates the combination encorafenib-cetuximab associated with chemotherapy. Similarly, a phase II trial is evaluating the combination vemurafenib-cetuximab with FOLFIRI (NCT04607421, NCT03727763) [68]. Time will let us know if these results will change standard of care. Ongoing clinical trials are resulted in Table 2.
Table 2. Ongoing clinical trials in mutated BRAF V600E mCRC.
Conclusion
Patients with mutated BRAF V600E mCRC harbour poor prognosis and short survival. Immunotherapy, the combination of BRAF and EGFR inhibitors and the combination of chemotherapy with angiogenic inhibitors have managed to prolong the survival of this population. Choosing the right treatment depends on each patient’s clinical and molecular features. The study of BRAF signalling pathway allowed the discovery of novel agents that are being studied right now. The present and future of mutated BRAF V600E mCRC treatment may be seen with hope.
Conflict of interest
The authors declare that they have no financial or non-financial conflicts of interest.
Author contributions
Rodrigo Motta Guerrero, Miguel Sotelo Lezama and Alejandro Figueroa Torrejon made the contributions to the conception and design of the work. Rodrigo Motta Guerrero, Veronica Arnao Labajos, Sophia Lozano Ballena and Carlos Aliaga Macha made the first draft. Alejandro Figueroa Torrejon, Paola Montenegro Beltran and Cristian Pacheco Roman critically reviewed the draft for intellectual content and made significant changes. All authors approved the final version of the manuscript.
Acknowledgments
None.
Funding statement
No funding was required for this article.
References
1. Sung H, Ferlay J, and Siegel RL, et al (2021) Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries CA: Cancer J Clin 71 209–249 PMID: 33538338
2. Bokemeyer C, Bondarenko I, and Hartmann JT, et al (2011) Efficacy according to biomarker status of cetuximab plus FOLFOX-4 as first-line treatment for metastatic colorectal cancer: the OPUS study Ann Oncol 22 1535–1546 https://doi.org/10.1093/annonc/mdq632 PMID: 21228335
3. Bokemeyer C, Van Cutsem E, and Rougier P, et al (2012) Addition of cetuximab to chemotherapy as first-line treatment for KRAS wildtype metastatic colorectal cancer: pooled analysis of the CRYSTAL and OPUS randomised clinical trials Eur J Cancer 48 1466–1475 https://doi.org/10.1016/j.ejca.2012.02.057 PMID: 22446022
4. Douillard JY, Siena S, and Cassidy J, et al (2014) Final results from PRIME: randomized phase III study of panitumumab with FOLFOX4 for firstline treatment of metastatic colorectal cancer Ann Oncol 25 1346–1355 https://doi.org/10.1093/annonc/mdu141 PMID: 24718886
5. Rivera F, Karthaus M, and Hecht JR, et al (2017) Final analysis of the randomised PEAK trial: overall survival and tumour responses during firstline treatment with mFOLFOX6 plus either panitumumab or bevacizumab in patients with metastatic colorectal carcinoma Int J Colorectal Dis 32 1179–1190 https://doi.org/10.1007/s00384-017-2800-1 PMID: 28424871 PMCID: 5522523
6. Cremolini C, Schirripa M, and Antoniotti C, et al (2015) First-line chemotherapy for mCRC a review and evidence-based algorithm Nat Rev Clin Oncol 12 607–619 https://doi.org/10.1038/nrclinonc.2015.129 PMID: 26215044
7. Motta R, Cabezas-Camarero S, and Torres-Mattos C, et al (2021) Immunotherapy in microsatellite instability metastatic colorectal cancer: current status and future perspectives J Clin Transl Res 7(4) 511–522 PMID: 34541365 PMCID: 8445628
8. Motta R, Cabezas-Camarero S, and Torres-Mattos C, et al (2021) Personalizing first-line treatment in advanced colorectal cancer: present status and future perspectives J Clin Transl Res 7(6) 771–785
9. Domingo E, Niessen RC, and Oliveira C, et al (2005) BRAF-V600E is not involved in the colorectal tumorigenesis of HNPCC in patients with functional MLH1 and MSH2 genes Oncogene 24(24) 3995–3998 https://doi.org/10.1038/sj.onc.1208569 PMID: 15782118
10. Tran B, Kopetz S, and Tie J, et al (2011) Impact of BRAF mutation and microsatellite instability on the pattern of metastatic spread and prognosis in metastatic colorectal cancer Cancer 117 4623–4632 https://doi.org/10.1002/cncr.26086 PMID: 21456008 PMCID: 4257471
11. Cremolini C, Di Bartolomeo M, and Amatu A, et al (2015) BRAF codons 594 and 596 mutations identify a new molecular subtype of metastatic colorectal cancer at favorable prognosis Ann Oncol 26(10) 2092–2097 https://doi.org/10.1093/annonc/mdv290 PMID: 26153495
12. Fanelli GN, Dal Pozzo CA, and Depetris I, et al (2020) The heterogeneous clinical and pathological landscapes of metastatic Braf-mutated colorectal cancer Cancer Cell Int 20 30 https://doi.org/10.1186/s12935-020-1117-2 PMID: 32015690 PMCID: 6990491
13. Taieb J, Lapeyre-Prost A, and Laurent Puig P, et al (2019) Exploring the best treatment options for BRAF-mutant metastatic colon cancer Br J Cancer 121 434–442 https://doi.org/10.1038/s41416-019-0526-2 PMID: 31353365 PMCID: 6738120
14. Śmiech M, Leszczyński P, and Kono H, et al (2020) Emerging BRAF mutations in cancer progression and their possible effects on transcriptional networks Genes 11(11) 1342 https://doi.org/10.3390/genes11111342 PMID: 33198372 PMCID: 7697059
15. Zaman A, Wu W, and Bivona TG (2019) Targeting oncogenic BRAF: past, present, and future Cancers 11(8) 1197 https://doi.org/10.3390/cancers11081197 PMID: 31426419 PMCID: 6721448
16. Nakayama I, Hirota T, and Shinozaki E (2020) BRAF mutation in colorectal cancers: from prognostic marker to targetable mutation Cancers (Basel) 12(11) 3236 https://doi.org/10.3390/cancers12113236 PMID: 33152998 PMCID: 7694028
17. Barras D, Missiaglia E, and Wirapati P, et al (2017) Braf v600e mutant colorectal cancer subtypes based on gene expression Clin Cancer Res 23 104–115 https://doi.org/10.1158/1078-0432.CCR-16-0140
18. Grassi E, Corbelli J, and Papiani G, et al (2021) Current therapeutic strategies in BRAF-mutant metastatic colorectal cancer Front Oncol 11 601722 https://doi.org/10.3389/fonc.2021.601722 PMID: 34249672 PMCID: 8262685
19. Caputo F, Santini C, and Bardasi C, et al (2019) BRAF-mutated colorectal cancer: clinical and molecular insights Int J Mol Sci 20(21) 5369 https://doi.org/10.3390/ijms20215369 PMID: 31661924 PMCID: 6861966
20. Stintzing S, Miller-Phillips L, and Modest DP, et al (2017) Impact of BRAF and RAS mutations on first-line efficacy of FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab: analysis of the FIRE-3 (AIO KRK-0306) study Eur J Cancer 79 50–60 https://doi.org/10.1016/j.ejca.2017.03.023 PMID: 28463756
21. Price TJ, Hardingham JE, and Lee CK, et al (2011) Impact of KRAS and BRAF gene mutation status on outcomes from the phase III AGITG MAX Trial of capecitabine alone or in combination with bevacizumab and mitomycin in advanced colorectal cancer J Clin Oncol 29(19) 2675–2682 https://doi.org/10.1200/JCO.2010.34.5520 PMID: 21646616
22. Grothey A, Fakih M, and Tabernero J (2021) Management of BRAF-mutant metastatic colorectal cancer: a review of treatment options and evidence-based guidelines Ann Oncol 32(8) 959–967 https://doi.org/10.1016/j.annonc.2021.03.206 PMID: 33836264
23. Loupakis F, Cremolini C, and Salvatore L, et al (2014) FOLFOXIRI plus bevacizumab as first-line treatment in BRAF mutant metastatic colorectal cancer Eur J Cancer 50(1) 57–63 https://doi.org/10.1016/j.ejca.2013.08.024
24. Masi G, Loupakis F, and Salvatore L, et al (2010) Bevacizumab with FOLFOXIRI (irinotecan, oxaliplatin, fluorouracil, and folinate) as first-line treatment for metastatic colorectal cancer: a phase 2 trial Lancet Oncol 11(9) 845–852 https://doi.org/10.1016/S1470-2045(10)70175-3 PMID: 20702138
25. Cremolini C, Loupakis F, and Antoniotti C, et al (2015) FOLFOXIRI plus bevacizumab versus FOLFIRI plus bevacizumab as first-line treatment of patients with metastatic colorectal cancer: updated overall survival and molecular subgroup analyses of the open-label, phase 3 TRIBE study Lancet Oncol 16(13) 1306–1315 https://doi.org/10.1016/S1470-2045(15)00122-9 PMID: 26338525
26. Cremolini C, Antoniotti C, and Rossini D, et al (2020) Upfront FOLFOXIRI plus bevacizumab and reintroduction after progression versus mFOLFOX6 plus bevacizumab followed by FOLFIRI plus bevacizumab in the treatment of patients with metastatic colorectal cancer (TRIBE2): a multicentre, open-label, phase 3, randomised, controlled trial Lancet Oncol 21(4) 497– https://doi.org/10.1016/S1470-2045(19)30862-9 PMID: 32164906
27. Zhou M, Yu P, and Hernick Davin DB, et al (2017) Is FOLFOXIRI alone or combined with targeted therapy administered as first-line treatment a reasonable choice for most patients with mCRC? Systematic review and network meta-analysis Oncotarget 8(37) 62339–62348 https://doi.org/10.18632/oncotarget.17725 PMID: 28977949 PMCID: 5617509
28. Cremolini C, Antoniotti C, and Stein A, et al (2020) Individual patient data meta-analysis of FOLFOXIRI plus bevacizumab versus doublets plus bevacizumab as initial therapy of unresectable metastatic colorectal cancer J Clin Oncol https://doi.org/10.1200/JCO.20.01225 PMID: 32816630
29. Orlandi A, Calegari A, and Inno A, et al (2015) BRAF in metastatic colorectal cancer: the future starts now Pharmacogenomics 16(18) 2069–2081 https://doi.org/10.2217/pgs.15.140 PMID: 26615988
30. Geissler M, Martens UM, and Knorrenschield R, et al (2017) 475OmFOLFOXIRI + panitumumab versus FOLFOXIRI as first-line treatment in patients with RAS wild-type metastatic colorectal cancer m(CRC): a randomized phase II VOLFI trial of the AIO (AIO-KRK0109) Ann Oncol 28(Suppl 5) Abstract 475O https://doi.org/10.1093/annonc/mdx393.002
31. Geissler M, Riera-Knorrenschild J, and Martens UM, et al (2019) Final results and OS of the randomized phase II VOLFI trial (AIO- KRK0109): mFOLFOXIRI + panitumumab versus FOLFOXIRI as first-line treatment in patients with RAS wild- type metastatic colorectal cancer (mCRC) J Clin Oncol 36(15_suppl) 3509–3509 https://doi.org/10.1200/JCO.2018.36.15_suppl.3509
32. Le DT, Kim TW, and Van Cutsem E, et al (2020) Phase II open-label study of pembrolizumab in treatment-refractory, microsatellite instability-high/mismatch repair-deficient metastatic colorectal cancer: KEYNOTE-164 J Clin Oncol 38(1) 11–19 https://doi.org/10.1200/JCO.19.02107 PMCID: 7031958
33. Overman MJ, McDermott R, and Leach JL, et al (2017) Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study Lancet Oncol 18(9) 1182–1191 Erratum in: Lancet Oncol 18(9) e510 https://doi.org/10.1016/S1470-2045(17)30422-9 PMID: 28734759 PMCID: 6207072
34. Overman MJ, Lonardi S, and Wong KYM, et al (2018) Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer J Clin Oncol 36(8) 773–779 https://doi.org/10.1200/JCO.2017.76.9901 PMID: 29355075
35. Lenz HJ, Lonardi S, and Zagonel V, et al (2020) Nivolumab plus low-dose ipilimumab as first-line therapy in microsatellite instability-high/DNA mismatch repair deficient metastatic colorectal cancer: clinical update J Clin Oncol 38(4_suppl) 11 https://doi.org/10.1200/JCO.2020.38.4_suppl.11
36. André T, Shiu KK, and Kim TW, et al (2020) Pembrolizumab in microsatellite-instability-high advanced colorectal cancer N Engl J Med 383(23) 2207–2218 https://doi.org/10.1056/NEJMoa2017699 PMID: 33264544
37. Shiu KK, Andre T, and Kim TW, et al (2021) KEYNOTE-177: phase III randomized study of pembrolizumab versus chemotherapy for microsatellite instability-high advanced colorectal cancer J Clin Oncol 39(3_suppl) 6 https://doi.org/10.1200/JCO.2021.39.3_suppl.6
38. Park R, Lopes L, and Lee S, et al (2021) The prognostic and predictive impact of BRAF mutations in deficient mismatch repair/microsatellite instability-high colorectal cancer: systematic review/meta-analysis Future Oncol 17(31) 4221–4231 https://doi.org/10.2217/fon-2021-0552 PMID: 34323124
39. Hyman DM, Puzanov I, and Subbiah V, et al (2015) Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations N Engl J Med 373(8) 726–736 Erratum in: N Engl J Med 379(16) 1585 https://doi.org/10.1056/NEJMoa1502309 PMID: 26287849 PMCID: 4971773
40. Kopetz S, Desai J, and Chan E, et al (2015) Phase II pilot study of vemurafenib in patients with metastatic BRAF-mutated colorectal cancer J Clin Oncol 33(34) 4032–4038 https://doi.org/10.1200/JCO.2015.63.2497 PMID: 26460303 PMCID: 4669589
41. Gomez-Roca CA, Delord J, and Robert C, et al (2014) Encorafenib (LGX818), an oral BRAF inhibitor, in patients (pts)with BRAF V600E metastatic colorectal cancer (MCRC): results of dose expansion in an open-label phase 1 study Ann Oncol 25(suppl 4) iv167–iv209 https://doi.org/10.1093/annonc/mdu333.38
42. Corcoran RB, Atreya CE, and Falchook GS, et al (2015) Combined BRAF and MEK inhibition with dabrafenib and trametinib in BRAF V600-mutant colorectal cancer J Clin Oncol 33(34) 4023–4031 https://doi.org/10.1200/JCO.2015.63.2471 PMID: 26392102 PMCID: 4669588
43. Huijberts SCFA, Boelens MC, and Bernards R, et al (2021) Mutational profiles associated with resistance in patients with BRAFV600E mutant colorectal cancer treated with cetuximab and encorafenib +/- binimetinib or alpelisib Br J Cancer 124(1) 176–182 https://doi.org/10.1038/s41416-020-01147-2 PMCID: 7782586
44. van Geel RMJM, Tabernero J, and Elez E, et al (2017) A phase Ib dose-escalation study of encorafenib and cetuximab with or without alpelisib in metastatic BRAF-mutant colorectal cancer Cancer Discov 7(6) 610–619 https://doi.org/10.1158/2159-8290.CD-16-0795 PMID: 28363909 PMCID: 5546207
45. Corcoran RB, Ebi H, and Turke AB, et al (2012) EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib Cancer Discov 2(3) 227–235 https://doi.org/10.1158/2159-8290.CD-11-0341 PMID: 22448344 PMCID: 3308191
46. Corcoran RB, André T, and Atreya CE, et al (2018) Combined BRAF, EGFR, and MEK inhibition in patients with BRAFV600E-mutant colorectal cancer Cancer Discov 8 428–443 https://doi.org/10.1158/2159-8290.CD-17-1226 PMID: 29431699 PMCID: 5882509
47. Kopetz S, Grothey A, and Yaeger R, et al (2019) Encorafenib, binimetinib, and cetuximab in BRAF V600E-mutated colorectal cancer N Engl J Med 381(17) 1632–1643 https://doi.org/10.1056/NEJMoa1908075 PMID: 31566309
48. Tabernero J, Grothey A, and Van Cutsem E, et al (2021) Encorafenib plus cetuximab as a new standard of care for previously treated BRAF V600E-mutant metastatic colorectal cancer: updated survival results and subgroup analyses from the BEACON study J Clin Oncol 39(4) 273–284 https://doi.org/10.1200/JCO.20.02088 PMID: 33503393 PMCID: 8078423
49. Grothey A, Tabernero J, and Taieb J, et al (2020) LBA-5 ANCHOR CRC: a single-arm, phase 2 study of encorafenib, binimetinib plus cetuximab in previously untreated BRAF V600E-mutant metastatic colorectal cancer Ann Oncol 31 S242–S243 https://doi.org/10.1016/j.annonc.2020.04.080
50. Kopetz S, Guthrie KA, and Morris VK, et al (2021) Randomized trial of irinotecan and cetuximab with or without vemurafenib in BRAF-mutant metastatic colorectal cancer (SWOG S1406) J Clin Oncol 39(4) 285–294 https://doi.org/10.1200/JCO.20.01994 PMCID: 8462593
51. Callebout E, Ribeiro SM, and Laurent S, et al (2019) Long term response on regorafenib in non-V600E BRAF mutated colon cancer: a case report BMC Cancer 19(1) 567 https://doi.org/10.1186/s12885-019-5763-5 PMID: 31185985 PMCID: 6560823
52. Dankner M (2018) Targeted therapy for colorectal cancers with non-V600 BRAF mutations: perspectives for precision oncology JCO Precis Oncol 2 1–12 PMID: 35135170
53. Molina-Cerrillo J, San Román M, and Pozas J, et al (2020) BRAF mutated colorectal cancer: new treatment approaches Cancers (Basel) 12(6) 1571 https://doi.org/10.3390/cancers12061571 PMID: 32545884 PMCID: 7353017
54. Gómez-España MA, Gallego J, and González-Flores E, et al (2019) SEOM clinical guidelines for diagnosis and treatment of metastatic colorectal cancer (2018) Clin Transl Oncol 21(1) 46–54 https://doi.org/10.1007/s12094-018-02002-w
55. Van Cutsem E, Cervantes A, and Adam R, et al (2016) ESMO consensus guidelines for the management of patients with metastatic colorectal cancer Ann Oncol 27(8) 1386–1422 https://doi.org/10.1093/annonc/mdw235 PMID: 27380959
56. Yoshino T, Arnold D, and Taniguchi H, et al (2018) Pan-Asian adapted ESMO consensus guidelines for the management of patients with metastatic colorectal cancer: a JSMO-ESMO initiative endorsed by CSCO, KACO, MOS, SSO and TOS Ann Oncol 29(1) 44–70 https://doi.org/10.1093/annonc/mdx738
57. Hashiguchi Y, Muro K, and Saito Y, et al (2020) Japanese Society for cancer of the colon and rectum (JSCCR) guidelines 2019 for the treatment of colorectal cancer Int J Clin Oncol 25(1) 1–42 https://doi.org/10.1007/s10147-019-01485-z PMCID: 6946738
58. National Comprehensive Cancer Network (NCCN) (2022) Colon Cancer. Version 1. 2022 (National Comprehensive Cancer Network)
59. Pembrolizumab for untreated metastatic colorectal cancer with high microsatellite instability or mismatch repair deficiency (2021) (National Institute for Health and Care Excellence (NICE))
60. Marcus L, Lemery SJ, and Keegan P, et al (2019) FDA approval summary: pembrolizumab for the treatment of microsatellite instability-high solid tumors Clin Cancer Res 25(13) 3753–3758 https://doi.org/10.1158/1078-0432.CCR-18-4070 PMID: 30787022
61. Boilève A and Samalin E (2020) Nouvelle AMM: encorafenib–cancers colorectaux métastatiques mutés BRAF V600E après chimiothérapie systémique [New drug approval: encorafenib-metastatic colorectal cancers with BRAF V600E mutation after systemic chemotherapy] Bull Cancer 107(11) 1086–1088 [French] https://doi.org/10.1016/j.bulcan.2020.08.012 PMID: 33046237
62. Trullas A, Delgado J, and Koenig J, et al (2021) The EMA assessment of encorafenib in combination with cetuximab for the treatment of adult patients with metastatic colorectal carcinoma harbouring the BRAFV600E mutation who have received prior therapy ESMO Open 6(1) 100031 https://doi.org/10.1016/j.esmoop.2020.100031 PMID: 33422765 PMCID: 7809377
63. Encorafenib Plus Cetuximab for Previously Treated BRAF V600E Mutation-Positive Metastatic Colorectal Cancer. Technology Appraisal Guidance (2021) [www.nice.org.uk/guidance/ta668]
64. ClinicalTrials.gov [Internet] Identifier NCT04044430, Phase I/II Trial of Encorafenib, Binimetinib, and Nivolumab in Microsatellite Stable BRAF V600E Metastatic Colorectal Cancer (National Library of Medicine) [https://ClinicalTrials.gov/ct2/show/record/NCT04044430?term=braf&cond=Colon+Cancer&draw=2]
65. Damato A, Berselli A, and Iachetta F, et al (2021) Preliminary safety analysis of phase II open-label NIVACOR trial (GOIRC-03-2018) in patients with advanced colorectal cancer RAS or BRAF mutated J Clin Oncol 39(3_suppl) 37 https://doi.org/10.1200/JCO.2021.39.3_suppl.37
66. Sharma A, Madhunapantula SV, and Gowda R, et al (2013) Identification of aurora kinase B and Wee1-like protein kinase as downstream targets of V600EB-RAF in melanoma Am J Pathol 182 1151–1162 https://doi.org/10.1016/j.ajpath.2012.12.019 PMID: 23416158 PMCID: 3620396
67. ClinicalTrials.gov NCT02278133_study of WNT974 in combination with LGX818 and cetuximab in patients with BRAF-mutant metastatic colorectal cancer (mCRC) and Wnt pathway mutations [https://ClinicalTrials.gov/ct2/show/NCT02278133]
68. Kopetz S, Grothey A, and Yaeger R, et al (2021) BREAKWATER: randomized phase 3 study of encorafenib (enco) + cetuximab (cetux) ± chemotherapy for first-line (1L) treatment (tx) of BRAF V600E-mutant (BRAFV600E) metastatic colorectal cancer (mCRC) J Clin Oncol 39(15_suppl) TPS3619-TPS3619 https://doi.org/10.1200/JCO.2021.39.15_suppl.TPS3619





