miRNAs and resistance to EGFR—TKIs in EGFR-mutant non-small cell lung cancer: beyond ‘traditional mechanisms’ of resistance
Biagio Ricciuti1, Carmen Mecca2, Matteo Cenci1, Giulia Costanza Leonardi1, Lorenzo Perrone1, Clelia Mencaroni1, Lucio Crinò1, Francesco Grignani3, Sara Baglivo1, Rita Chiari1, Angelo Sidoni3, Luca Paglialunga1, Maria Francesca Currà1, Emanuele Murano1, Vincenzo Minotti1 and Giulio Metro1
1Medical Oncology, Santa Maria della Misericordia Hospital, Azienda Ospedaliera di Perugia, Perugia 06156, Italy
2Department of Experimental Medicine, University of Perugia, Perugia 06156, Italy
3Department of Clinical and Experimental Medicine, Division of Pathology, University of Perugia, Perugia 06156, Italy
Correspondence to: Giulio Metro. Email: giulio.metro@yahoo.com
Abstract
Epidermal growth factor receptor (EGFR)-tyrosine kinase inhibitors (TKIs) have dramatically changed the prognosis of advanced non-small cell lung cancers (NSCLCs) that harbour specific EGFR activating mutations. However, the efficacy of an EGFR-TKI is limited by the onset of acquired resistance, usually within one year, in virtually all treated patients. Moreover, a small percentage of EGFR-mutant NSCLCs do not respond to an EGFR-TKI, thus displaying primary resistance. At the present time, several mechanisms of either primary and acquired resistance have been elucidated, and new drugs are currently under preclinical and clinical development in order to overcome resistance to treatment. Nevertheless, there still remains much to be thoroughly investigated, as so far research has mainly focused on the role of proteincoding genes involved in resistance to EGFR-TKIs. On the other hand, in line with the data underscoring the relevance of non-coding RNAs in the pathogenesis of lung cancer and modulation of response to systemic therapies, microRNAs (miRNAs) have been supposed to play an important role in resistance to EGFR-TKIs. The aim of this review is to briefly summarise the existing relationship between miRNAs and resistance to EGFR-TKIs, and also focusing on the possible clinical applications of miRNAs in reverting and overcoming such resistance.
Keywords: EGFR mutation, EGFR-TKI, NSCLC, miRNAs, resistance
Copyright: © the authors; licensee ecancermedicalscience. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Published: 02/09/2015; Received: 11/05/2015
Introduction
Lung cancer is the leading cause of cancer-related deaths worldwide, representing 27.2% of all cancer deaths [1]. As many other neoplasms, lung cancer has been recognised as a group of different diseases, each with its own clinical and biological features. On one hand, non-small cell lung cancer (NSCLC), which accounts for approximately 80% of all lung cancers, on the other, small cell lung cancer (SCLC), which accounts for the rest of cases [2]. Even though enormous improvements have been made in the systemic treatment of advanced NSCLC, particularly for the non-squamous subgroup, the prognosis of these patients is still extremely poor, five-year survival being 5% and 1%, respectively [3]. The reason for this poor outcome should be attributed, at least in part, to the fact that platinum-based chemotherapy represents the standard of care for the majority of patients with advanced disease, irrespective of tumour biology, which does not take into account the intra- and inter-individual heterogeneity of each lung cancer case [4]. Nevertheless, recent breakthroughs in the understanding of NSCLC biology have led to the discovery of specific genetic alterations, such as EGFR mutations, anaplastic lymphoma kinase (ALK), and ROS1 gene rearrangements, each of which identifies a distinct disease entity which has been termed ‘oncogene addicted’ in order to reflect its dependence on a single genetic ‘driver’ for proliferation and survival of cancer cells.
With regard to EGFR-mutant NSCLC, the introduction for therapeutic use of EGFR-TKIs, either reversible, first-generation (gefitinib, erlotinib) or irreversible, second-generation (afatinib) agents, has dramatically improved the prognosis of patients whose tumour harbours this specific genetic alteration (Table 1) [5].
In this paper, we will briefly summarise the most well known mechanisms of resistance to EGFR-TKIs. Also, our exploration will go beyond traditional mechanisms of resistance, particularly focusing on the ability of microRNAs (miRNAs) to regulate response to EGFR-TKIs. Finally, the possible clinical applications of miRNAs in escaping resistance to EGFR-TKIs will be discussed.
EGFR mutations and EGFR-TKIs in NSCLC
EGFR (ERBB1/HER1) is a member of a transmembrane receptors tyrosine kinase (TK) superfamily, which also includes ERBB2/HER2, ERBB3/HER3, and ERBB4/HER4 [19]. Similarly to other members, EGFR is characterised by an extracellular ligand-binding region, a single transmembrane region, and an intracytoplasmic region with TK activity [20]. When an activating somatic mutation occurs in the EGFR-TK domain, EGFR undergoes ligand-independent homo/hetero-dimerisation with another receptor of the same family. This leads to conformational changes in the three-dimensional structure of EGFR that promote ATP-mediated autophosphorylation of the TK domain, and subsequent receptor activation [21, 22]. As a consequence, multiple intracellular signalling cascades are implemented, such as RAS/RAF/MEK/ERK, JAK/STAT, and PI3K/AKT/mTOR, all of which result into intensification of pro-survival and anti-apoptotic signalling [19, 22].
Interestingly, although EGFR mutations are more commonly associated with specific clinicopathological features such as female gender, Asian ethnicity (where they can be found in up to 30% of advanced NSCLCs as opposed to 15% for the western population), non-smoking history, and adenocarcinoma histology [23], it is not possible to rule out the possibility of an EGFR mutation solely on the basis of the aforementioned characteristics. Also, only activating mutations that affect the TK domain (exons 18→21) are those who predict exquisite sensitivity to treatment with an EGFR-TKI, since EGFR-TKIs and ATP compete for binding to the same pocket on the EGFR-TK domain [24].
EGFR activating mutations include in frame deletions, in frame duplications/insertions, and point mutations [Murray 2008]. Specifically, the most represented and better characterised are the so called classic mutations, which include in frame deletions in exon 19 in correspondence of the LeuArgGluAla sequence (E746-A750), and the exon 21 point mutation Leu858Arg (L858R), together representing 85%–90% of all EGFR mutations in NSCLC [25]. On the other hand, a number of ‘uncommon’ mutations has been reported, such as G719X in exon 18 (G719C, G719S, G719A), L861Q in exon 21, and S768I in exon 20, whose predictive role to treatment with an EGFR-TKI is much less defined. Nevertheless, a growing body of evidence seems to confirm a positive predictive role of these uncommon mutations to treatment with an EGFR-TKI [26–28], although to a less extent than classic mutations [29]. Conversely, exon 20 in frame insertion as well as a de novo T790M point mutation in exon 20 are associated with primary resistance to EGFR-TKIs [26–28].
Table 1. Phase III studies in which gefitinib, erlotinib, or afatinib were compared with platinum-based chemotherapy as first-line treatment of patients with advanced NSCLC selected according to the presence of EGFR mutation.
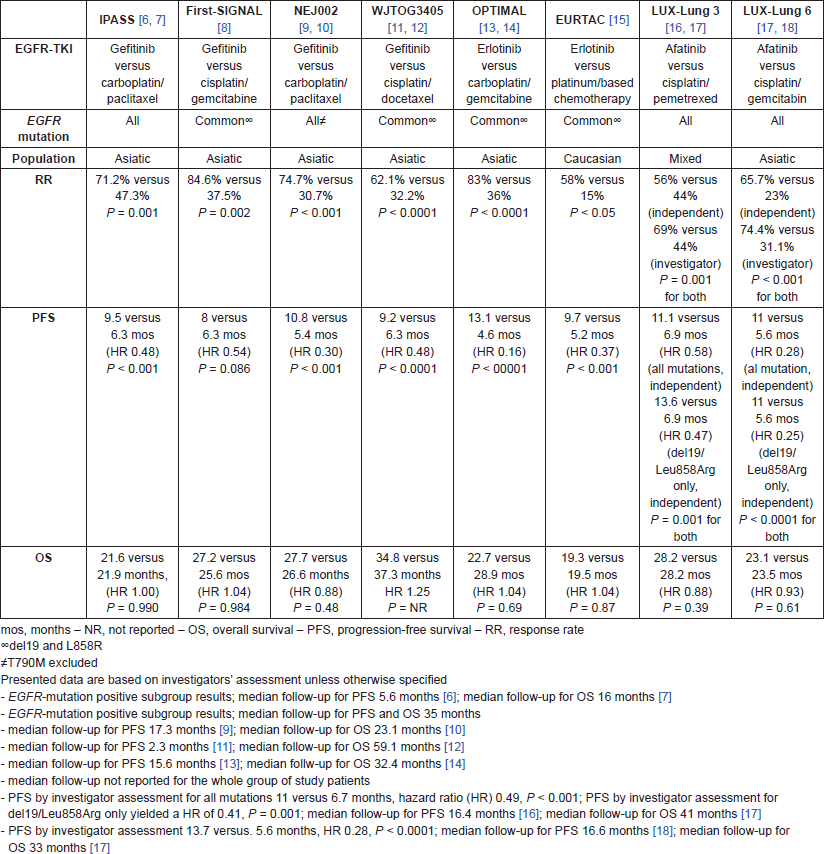
Recently, six phase III randomised trials comparing either a reversible (gefitinib or erlotinib) or irreversible (afatinib) EGFR-TKI versus platinum-based chemotherapy in patients with untreated EGFR-mutant advanced NSCLC showed superiority for the EGFR-TKI in terms of response rate (RR) and progression free survival (PFS) (Table 1) [6–18], as well as quality of life (QOL) [6, 13, 30]. These results corroborated further those of the earlier IPASS and first-SIGNAL trials, in which gefitinib was compared head-to-head with platinum-based chemotherapy in a population enriched for the presence of an EGFR mutation, such as East Asian and never/light smoker patients (Table 1) [6, 8]. In both studies gefitinib demonstrated a significant increase in RR and PFS only for the EGFR-mutant subgroup, whereas EGFR wild type patients were found to benefit more from standard chemotherapy than gefitinib. Importantly, although first-line afatinib significantly improved OS compared to chemotherapy in patients with EGFR Del19 mutations in two randomised trials, LUX-Lung 3 and LUX-Lung 6, there was no significant difference in OS of patients with EGFR L858R mutation treated with afatinib compared to chemotherapy, both individually and in pooled data [31]. These findings suggest that patients harbouring EGFR Del19 and L858R mutations may represent two separate populations and further studies are required to address this issue.
Unfortunately, despite the impressive activity of EGFR-TKIs in EGFR-mutant NSCLCs, virtually all patients would undergo resistance to treatment, usually within one year of treatment initiation. Against this background, it is of primary importance to understand the molecular mechanisms underlying resistance to EGFR-TKIs in order to develop novel therapeutic strategies aimed at improving further the prognosis of EGFR-mutant NSCLCs.
‘Traditional mechanisms’ of primary and acquired resistance to EGFR-TKIs
Currently, 20–30% of EGFR-mutant NSCLC patients do not undergo tumour shrinkage on treatment with an EGFR-TKI, thus identifying a primary resistant cohort. Furthermore, virtually all patients who have experienced either an objective response or prolonged disease stabilisation (≥ six months) on an EGFR-TKI will eventually develop resistance to the drug, which identifies the acquired resistant cohort [32].
With regard to primary resistance, exon 20 insertions have been invariably associated with lack of response to EGFR-TKIs, as suggested by numerous clinical studies [26–28, 33]. Also, de novo T790M point mutation in exon 20 identifies a group of patients who perform poorly on EGFR-TKIs. Although considered rather uncommon, highly sensitive method based on laser microdissection and peptide nucleic acid (PNA)-clamping polymerase chain reaction (PCR) recently unveiled an unexpected prevalence of de novo T790M substitution in patients with primary resistance to EGFR TKIs (2%–9%) [34–35]. This mutation, which is actually the major determinant of acquired resistance, is worthy of note and will be handled further along. Moreover, various alterations in the EGFR pathway may affect patients with NSCLC, thus conditioning response to EGFR-TKIs. More in detail, loss of phosphatase and tensin homolog PTEN expression, which is an onco-suppressor gene involved in the inhibition of PI3K/AKT/mTOR pathway, as well as PIK3CA activating mutations have both been reported in vitro as inducers of primary resistance to gefitinib and erlotinib through the blockade of EGFR-TKI-induced apoptosis in EGFR-mutant cell lines [34, 39]. In addition, BIM deletion polymorphism has been found to induce resistance to EGFR-TKIs [40]. BIM is a member of the BCL-2 pro-apoptotic proteins family, which is involved in Bax/Bak-mediated cytochrome c release and apoptosis. Of note, the relevance of BIM levels in influencing clinical response to EGFR-TKIs is supported by the results of EURTAC study, which randomised EGFR-mutant patients to erlotinib or platinum-based chemotherapy [15]. Interestingly, the erlotinibtreated patients who had low/intermediate mRNA levels of BIM expression experienced a lower PFS compared with those who had high levels of BIM expression [41]. Conversely, no differences in PFS were observed in the chemotherapy arm according to mRNA levels of BIM.
Currently, approximately 60% of cases of acquired resistance appear to be associated with the presence of a secondary missense mutation termed T790M. Such a mutation is characterised by the replacement of a threonine with a methionine at codon 790 of exon 20 of the EGFR gene, in a site that affects the catalytic adenosine 5’ triphosphate (ATP) binding pocket of the EGFR-TK domain [42]. That is because T790M mutation restores the binding affinity between the EGFR-TK domain and ATP, thus identifying what has been called the ‘gatekeeper’ mutation [43]. Of note, Maheswaran et al have shown that in some EGFR-mutation positive NSCLCs who develop T790M-mediated acquired resistance, this mutation could actually be detected at baseline in minor clones with the use of highly sensitive techniques, thus becoming dominant during exposure to EGFR-TKIs as a result of selective pressure induced by treatment with an EGFR-TKI [44]. Nevertheless, a few other EGFR secondary point mutations have been involved in acquired resistance to an EGFR-TKI, namely D716Y (exon 19), L747S (exon 19), and T854A (exon 21) [45–47]. In addition, acquired resistance might be because of the presence of other mutant signalling proteins (e.g. PI3CKA mutation, HER-2 amplification, BRAF mutation) as well as to the activation of EGFR signalling pathways via other aberrant molecules. Among the latter mechanism, the most relevant is certainly MET gene amplification, as it drives roughly 5–10% of cases of acquired resistance to EGFR-TKIs [42]. MET encodes for a transmembrane TK receptor implicated in acquired resistance through the activation of the PI3K/AKT/mTOR pathway via interaction with ERBB3 [48]. Not surprisingly, hepatocyte growth factor (HGF), which is the only known ligand of MET receptor, has also been implicated both in primary and acquired resistance to EGFR-TKIs [49–51]. Finally, in a minority of cases some other additional mechanism may lead to the development of acquired resistance such as phenotypic changes in the tumour. This can be because of SCLC transformation as well as to epithelial-to-mesenchymal transition (EMT) [52–53]. Of note, cells displaying SCLC features still harbour a drug-sensitive EGFR mutation. On the other hand, an increasing body of evidence describes the molecular machinery involved in EMT, which includes a variety of phenotypic and molecular changes (e.g. loss of tight junction and cell adhesion, induction of a mesenchymal phenotype) responsible for the increased aggressiveness and invasiveness of the tumour [54]. Cells with EMT are associated with loss of E-cadherin and β-catenin, though they gain the expression of vimentin and N-cadherin. At a molecular level, NOTCH-1 upregulation, transforming growth factor TGF-β overexpression, activation of MET and AXL pathways have been identified as major determinants of EMT-induced acquired resistance to EGFR-TKIs [55–58]. Even though EGFR mutations are largely homogeneous, mechanisms of resistance are more heterogeneous. Multiple mechanisms of resistance can be detected in a single tumour specimen, and different mechanisms of resistance can emerge from different tumour sites in the same patient, indicating the polyclonal nature of TKIs resistance. For instance, by examining various progressive lesions in an autopsy case from a patient with acquired resistance to EGFR TKI, Balak and colleagues revealed that the seven different sites all harboured the exon 19 deletion, conversely the T790M mutation was found in six out of the seven sites, but was not detected in the brain metastasis, despite the use of highly sensitive procedure [45]. In addition, a recent large series study designed to assess prospectively the frequency of various mechanism of resistance in 155 patients revealed that 98 had a second-site EGFR T790M mutations (63%) and four had small cell transformation, whereas MET and HER-2 amplification were reported in four patients and three patients respectively. Even though acquired mutations in PIK3CA, AKT1, BRAF, ERBB2, KRAS, MEK1, and NRAS were not detected, the coexistence of different mechanisms of acquired resistance was observed in 4% of patients [59].
miRNAs and resistance to EGFR-TKIs
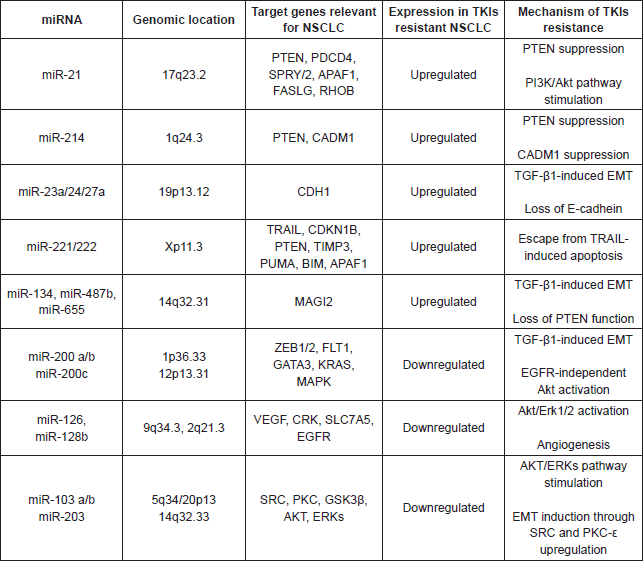
MiRNAs represent a class of 18–25 nucleotides in length, single stranded, endogenous and evolutionarily conserved small non-coding RNAs. They bind protein-coding mRNAs subsequently leading to mRNAs degradation, storage, and translational inhibition [60]. Although miRNAs account for barely 1%–2% of the human genome, they actually regulate and supervise the activity of roughly 50% of all protein-coding genes [61]. Currently, a number of miRNAs has been described which may have a specific role in lung cancer pathogenesis, biological and clinical disease behaviour as well as in modulating response to anticancer treatments, particularly EGFR-TKIs (Table 2) [60–63].
Located on chromosome 17q23.1, miR-21 is one of the most investigated miRNAs. Several studies reported its upregulation in NSCLC, and delineated its involvement in promoting lung cancer cell growth and invasion (Figure 1) [64–66]. Recently, Li et al demonstrated that miR-21 expression levels are higher in the EGFR-mutant (delE746-A750) and EGFR-TKI resistant NSCLC cell line PC9GR, inversely correlating with PTEN and PDCD4 expression [67]. On the other hand, miR-21 was significantly associated with the activation of the PI3K/Akt pathway. Notably, the same investigators found that miR-21 serum levels in EGFR-mutant NSCLC patients treated with an EGFR-TKI were significantly higher at the time of the onset of acquired resistance compared with baseline [67]. Additionally, in human EGFR-mutant NSCLC cell lines, miR-21 has been found to downregulate PTEN expression, a mechanism that could help promote lung carcinogenesis. MiR-21 expression has also been shown to associate with poor response and shorter overall survival (OS) in patients undergoing treatment with an EGFR-TKI [68]. Consistently, miR-21 overexpression decreased gefitinib sensitivity through PTEN downregulation and AKT/ERK pathway activation, whereas miR-21 knockdown restored gefitinib sensitivity through PTEN upregulation and AKT/ERK repression.
Table 2. Main miRNAs involved in EGFR TKIs resistance.
Similarly to miR-21, also miR-214 has been involved in acquired resistance to gefitinib through PTEN and PI3K/AKT pathway. More in details, Wang et al obtained gefitinib resistant cell line HCC827/GR through the exposure of normal HCC827 cells (NSCLC cell line with 746E-750A EGFR gene deletion) to increasingly larger concentrations of gefitinib and subsequently found that miR-214 was significantly overexpressed in HCC827/GR, also inversely correlating with the levels of PTEN expression [69]. Furthermore, not only miR-214 knockdown restored PTEN mRNA levels, but also was associated with p-Akt inactivation. Taken together, these data suggest that both miR-21 and miR-214 drive acquired resistance to gefitinib through the downregulation of PTEN expression, and therefore activation of the PI3K/AKT pathway independently of EGFR.
As previously mentioned, EMT represents an important determinant of clinical response to treatment with an EGFR-TKI, which has attracted much of an interest in miRNAs involved in the EMT phenomenon. Among them, miR-23a, miR-24, and miR-27a is a miRNAs cluster located on chromosome 19p13.12 with profound oncogenic properties in several human cancers. Cao et al showed that miR-23a is induced by the TGF-β1/Smad pathway in EGFR-WT A549 lung adenocarcinoma cells with the EMT phenomenon, whereas miR-24 and miR-27a are induced by a Smad-independent mechanism in the same cell lines [70]. These findings suggest that the activation of the TGF-β pathway contributes to EMT also through miR-23a/24/27a upregulation. In support of these data, there is evidence that miR-23a overexpression strengthens EMT and suppresses E-cadherin expression, whereas miR-23a knockdown restores E-cadherin expression [70]. Therefore, since miR-23a overexpression regulates TGF-β1-induced EMT and consequently EMT-related acquired resistance to gefitinib, miR-23a may be a new therapeutic target in both EGFR WT and EGFR-mutant NSCLC patients resistant to EGFR-TKIs.
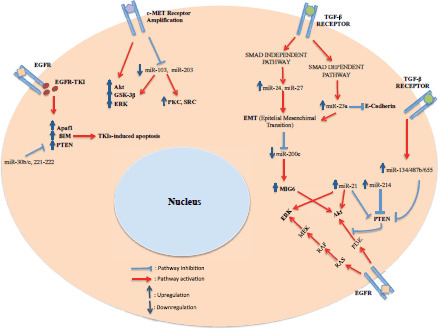
Figure 1. Schematic diagram of the relationship existing between miRNAs and resistance to EGFR-TKIs at a cellular level.
Still on EMT, the miR-200 family has been found to be downregulated during TGF-β1-induced EMT, leading to mitogen-inducible gene 6 (MIG6) upregulation. MIG6 is a negative regulator of EGFR and its overexpression in TGF-β1-induced EMT changes the molecular profile of NSCLC cells to an AKT-activated/EGFR-independent state. Izumchenko et al showed that expression levels of MIG6 and miR-200 were significantly correlated with EMT, as well as resistance to erlotinib in 25 cancer cell lines originating from different tissues; also the MIG6 mRNA/miR-200 ratio was inversely correlated with response to erlotinib in EGFR wild-type NSCLC patients [71]. In another study, Kitamura et al identified three other miRNAs, namely miR-134, miR-487b, and miR-655, all induced after exposure to TGF-β1 in lung adenocarcinoma cells with EMT [72]. Notably, the upregulation of these mi-RNAs results in loss of PTEN function, as one of the predicted targets of this cluster of miRNAs is the protein MAGI2, which is an essential scaffold protein for the proper functioning of PTEN. Overall, miR-134/487b/655 cluster promotes TGF-β1-induced EMT phenomenon, thus conditioning the development of ETM-driven acquired resistance to EGFR-TKIs [72].
Other authors examined EGFR and MET mediated changes of miRNAs in NSCLC cell lines. More in details, they showed that gefitinib treatment reduces miR-30b/c and miR-221/222 expression levels, consequently leading to the upregulation of APAF and BIM, which are both involved in determining EGFR-TKIs-induced apoptosis in sensitive EGFR-mutant, HCC827, and PC9 NSCLC cell lines [73–74]. Conversely, miR-30b/c and miR-221/222 downregulation was not observed after gefitinib treatment in HCC827/GR and PC9GR resistant clones of gefitinib hypersensitive EGFR exon 19 mutant HCC827 and PC9 NSCLC cell lines [74]. The same authors also showed that MET activation reduces expression levels of miR-103 and miR-203, which normally function as oncosuppressor miRNAs through the inhibition of SRC and PKC activity. Additionally, miR-103 and miR-203 were found to be significantly upregulated in MET knockdown Calu-1 NSCLC cells, whereas their overexpression was strongly related to reduced phosphorylation of Akt, GSK3β, and ERKs. This might suggest that MET amplification and overexpression may drive acquired resistance to EGFR-TKIs by enhancing AKT/ERKs pathway even through miR-103 and miR-203 regulation [74]. Taken together, these studies indicate that miR-30b/c and miR-221/222 upregulation in NSCLC determine resistance to EGFR-TKIs through the repression of APAF-1, BIM, and PTEN, while miR-103 and miR-203 induce apoptosis in EGFR-TKI resistant cells, also promoting mesenchymal to epithelial transition, by downregulating PKC and SRC.
miRNAs and new insights in overcoming resistance to EGFR-TKIs
Since miRNAs are involved in resistance to EGFR-TKIs, several studies explored the role of miRNAs in overcoming the mechanisms of resistance, with encouraging preclinical results. Zhou et al analysed miR-130a expression levels in H1975, A549, PC9 gefitinib-sensitive and -resistant NSCLC cell lines treated with different doses of gefitinib [75]. Interestingly, miR-130a levels were significantly lower in the gefitinib-resistant cell lines compared with the gefitinib-sensitive cell lines. Conversely, c-Met mRNA levels were higher in gefitinib-resistant than in gefitinib-sensitive cell lines. Even more importantly, it was demonstrated that transfection of miR-130a mimics could reverse resistance to gefitinib by downregulating c-Met protein levels via direct targeting of the c-Met 3’-UTR, which provides evidence for a new therapeutic strategy in reverting resistance to EGFR-TKIs [75]. Similarly, Acunzo et al highlighted the role of miRNAs in affecting c-Met [76]. More in details, they demonstrated that miR-130a downregulates c-Met, also reducing miR-221 and miR-222 expression. These two miRNAs have already been reported to be upregulated by c-Met activation/amplification, and are involved in cell proliferation, cell growth, and resistance to the TNF-related-apoptosis inducing ligand (TRAIL) protein in NSCLC [76–77]. Therefore, miR-130a overexpression could actually enhance apoptosis in NSCLC cells, and further studies should explore the possibility of a combined therapy with both miR-130a mimics and gefitinib, as both have been shown to revert resistance to TRAIL in human cancers [77].
More recently, Zhou et al also demonstrated that the forced expression of another miRNA, namely miR-34a, can inhibit cell growth and induce apoptosis in HGF-induced gefitinib-resistant HCC827 and PC-9 NSCLC cell lines via targeting c-Met [78]. Interestingly, the same study reported massive tumour regression with miR-34a plus gefitinib in mouse xenograft models of HGF-induced gefitinib-resistant tumours. The development of miR-34 as replacement therapy in EGFR-TKI resistant patients is a topic of growing interest, especially in light of its involvement in targeting c-Met and its oncogenic pathways [79]. Consistently, miR-34 has been found to inversely correlate with c-Met mRNA levels in lung, glioblastoma multiforme, and ovarian cancer [78–79]. Also, miR-34 involvement in the p53 tumour suppression pathway has been well described [80]. The importance of miR-34 in cancer development was further strengthened by Kasinski and Slack who evaluated the feasibility of the delivery of miR-34 lentivirus in order to prevent cancer initiation and progression in a therapeutically resistant mouse model of lung adenocarcinoma harbouring KRAS LSL-G12D/ and p53 LSL-R172H/ mutations [81]. Notably, miR-34 restoration induced cell-cycle arrest, apoptosis, and cellular senescence by targeting c-Met, c-myc, and the antiapoptotic protein BCL-2. These findings suggest that miR-34 replacement therapy may restore sensitivity to biological treatments in those patients in which the resistance to EGFR-TKIs is because of the amplification of MET. Moreover, miR-34, which is also able to suppress KRAS-driven cancer progression, could be of great benefit in patients whose acquired resistance depends on KRAS mutation. Finally, Zhao et al showed that miR-34 mimics enhance erlotinib sensitivity in NSCLC cell lines carrying both primary and acquired resistance to EGFR-TKIs [82], and two further studies confirmed that miR-34a might overcome resistance to EGFR-TKIs by targeting MET and AXL, which are both involved in erlotinib resistance [83–84].
More recently, Gao et al reported that miR-138-5 is deeply downregulated in gefitinib-resistant PC9 (harbouring delE746-A750) NSCLC cell lines, while its re-expression sensitise PC9/GR cells and another gefitinib-resistant EGFR-mutated NSCLC cell line, H1975 (harbouring both L858R/T790M mutations), to gefitinib. [85]. This phenomenon, which is mediated by G protein-coupled receptor 124, suggests that miR-138-5 replacement could be a further therapeutic approach in reversing resistance to EGFR-TKIs.
In addition, very attractive results have been obtained by Rai et al who reported that the injection of miR-7–expressing plasmid produces a dramatic growth arrest in either EGFR-mutated TKI-sensitive cell lines (PC9 and H3255) or EGFR TKI-resistant cell lines harbouring T790M mutation (R-PC9 and H1975) [86]. Importantly, no significant aberrant growth suppression was observed into A549 cells carrying wild-type EGFR and K-RAS mutations, thus highlighting that the oncogene addiction of EGFR plays a critical role in miR-7 efficacy.
As previously mentioned, EMT represents an important mechanism of acquired resistance to EGFR TKIs and different studies have shown that this process undergoes a precise regulation by specific miRNAs [54]. As a result, the possibility of reverting this process, leading to mesenchymal-to-epithelial transition through the use of miRNAs, was explored. To this regard, Lee et al found that miR-147 transfection in colon cancer (HCT116, SW480) and lung cancer (A-549) cell lines inhibited cell proliferation, cell migration, and reversed EMT to mesenchymal-to-epithelial transition through CDH1 upregulation and ZEB1 downregulation, thus markedly reestablishing gefitinib sensitivity [87].
Not surprisingly, also members of miRNAs biosynthetic machinery, such as the RNAse III enzyme Dicer, may be involved in TKIs resistance, as was proven by Chen et al who showed that Dicer expression levels are significantly higher in PC9/GR resistant cells than in PC9 cells and that Dicer knockdown restores gefitinib sensitivity in resistant cells through miR-30b/miR-30c and miR-221/miR-222 downregulation [88].
Conclusion and perspectives
Ever since their discovery, EGFR mutations have represented a major step forward in the management of NSCLC patients with this genetic alteration as they were associated with exquisite sensitivity to treatment with an EGFR-TKI. However, no patient was cured, and despite initial response in most of EGFR-mutant patients, resistance to treatment would eventually emerge. The drugs so far developed in order to overcome such resistance, including but not limited to T790M and c-Met inhibitors have been aimed at targeting only protein-coding genes implicated in resistance to treatment. Nevertheless, we also need to explore the universe of non-coding RNAs more thoroughly. In this setting, miRNAs appear to play a crucial role in regulating lung cancer carcinogenesis, sensitivity to chemotherapy, TKIs, as well as radiotherapy [89]. Moreover, their role as diagnostic, prognostic, and predictive biomarkers has been extensively investigated and it is an emerging topic in oncology. Certainly, their clinical use in restoring sensitivity to EGFR-TKIs is corroborated by robust preclinical evidence, which is an issue that needs to be investigated further in clinical trials. In order to achieve this, we must face the challenges regarding the use of miRNAs for NSCLC treatment (e.g. difficulties in identifying the appropriate methods of miRNAs delivery, determining the optimal therapeutic regimen, confirming their safety in humans), with the ultimate goal to benefit our patients.
Conflict of interests
The authors declare no conflict of interests.
Acknowledgments
Supported by the Italian Association for Cancer Research (AIRC).
References
1. Siegel R et al (2014) Cancer statistics, 2014 CA Cancer J Clin 64(1) 9–29 DOI: 10.3322/caac.21208 PMID: 24399786
2. Travis WD (2014) The 2015 WHO classification of lung tumors Pathologe 35(Suppl 2) 188 DOI: 10.1007/s00292-014-1974-3 PMID: 25394966
3. American Cancer Society Non-small cell lung cancer survival rates by stage Available at: http://cancer.org/cancer/lungcancernon smallcell/detailedguide/non-small-cell-lung-cancer-survival-rates [accessed January 1, 2015]
4. Reck M et al (2014) Metastatic non-small-cell lung cancer (NSCLC): ESMO clinical practice guidelines for diagnosis, treatment and follow-up Ann Oncol 25(suppl 3) iii27–39 DOI: 10.1093/annonc/mdu199 PMID: 25115305
5. Metro G and Crinò L (2012) Advances on EGFR mutation for lung cancer Transl Lung Cancer Res 1(1) 5–13 PMID: 25806150 PMCID: 4367583
6. Mok TS et al (2009) Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma N Engl J Med 361(10) 947–57 DOI: 10.1056/NEJMoa0810699 PMID: 19692680
7. Fukuoka M et al (2011) Biomarker analyses and final overall survival results from a phase III, randomized, open-label, first-line study of gefitinib versus carboplatin/paclitaxel in clinically selected patients with advanced non-small-cell lung cancer in Asia (IPASS) J Clin Oncol 29(21) 2866–74 DOI: 10.1200/JCO.2010.33.4235 PMID: 21670455
8. Han JY et al (2012) First-SIGNAL: first-line single-agent iressa versus gemcitabine and cisplatin trial in never-smokers with adenocarcinoma of the lung J Clin Oncol 30(10) 1122–8 DOI: 10.1200/JCO.2011.36.8456 PMID: 22370314
9. Maemondo M et al (2010) Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR N Engl J Med 362(25) 2380–8 DOI: 10.1056/NEJMoa0909530 PMID: 20573926
10. Inoue A et al (2013) Updated overall survival results from a randomized phase III trial comparing gefitinib with carboplatinpaclitaxel for chemo-naïve non-small cell lung cancer with sensitive EGFR gene mutations (NEJ002) Ann Oncol 24(1) 54–9 DOI: 10.1093/annonc/mds214
11. Mitsudomi T et al Gefitinib versus cisplatin plus docetaxel in patient with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial Lancet Oncol 11(2) 121–8 PMID: 20022809
12. Yoshioka H et al (2014) Final overall survival results of WJTOG 3405, a randomized phase 3 trial comparing gefitinib (G) with cisplatin plus docetaxel (CD) as the first-line treatment for patients with non-small cell lung cancer (NSCLC) harboring mutations of the epidermal growth factor receptor (EGFR) J Clin Oncol 32 (suppl 5; abstr 8117)
13. Zhou C et al (2011) Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study Lancet Oncol 12(8) 735–42 DOI: 10.1016/S1470-2045(11)70184-X PMID: 21783417
14. Zhou C et al (2012) Overall survival (OS) results from OPTIMAL (CTONG0802), a phase III trial of erlotinib (E) versus carboplatin plus gemcitabine (GC) as first-line treatment for Chinese patients with EGFR mutation-positive advanced non-small cell lung cancer (NSCLC) J Clin Oncol 30 (suppl; abstr 7520)
15. Rosell R et al (2012) Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial Lancet Oncol 13(3) 239–46 DOI: 10.1016/S1470-2045(11)70393-X PMID: 22285168
16. Sequist LV et al (2013) Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations J Clin Oncol 31(27) 3327–34 DOI: 10.1200/JCO.2012.44.2806 PMID: 23816960
17. Yang JC et al (2015) Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): analysis of overall survival data from two randomised, phase 3 trials Lancet Oncol 16(2) 141–51 DOI: 10.1016/S1470-2045(14)71173-8 PMID: 25589191
18. Wu YL et al (2014) Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial Lancet Oncol 15(2) 213–22 DOI: 10.1016/S1470-2045(13)70604-1 PMID: 24439929
19. Mitsudomi T and Yatabe Y (2010) Epidermal growth factor receptor in relation to tumor development: EGFR gene and cancer FEBS J 277(2) 301–8 DOI: 10.1111/j.1742-4658.2009.07448.x
20. Sibilia M et al (2007) The epidermal growth factor receptor: from development to tumorigenesis Differentiation 75(9) 770–87 DOI: 10.1111/j.1432-0436.2007.00238.x PMID: 17999740
21. Garrett TP et al (2002) Crystal structure of a truncated epidermal growth factor receptor extracellular domain bound to transforming growth factor alpha Cell 110(6) 763–73 DOI: 10.1016/S0092-8674(02)00940-6 PMID: 12297049
22. Burgess AW (2008) EGFR family: structure physiology signalling and therapeutic targets Growth Factors 26(5) 263–74 DOI: 10.1080/08977190802312844 PMID: 18800267
23. Mitsudomi T, Kosaka T and Yatabe Y (2006) Biological and clinical implications of EGFR mutations in lung cancer Int J Clin Oncol 11(3) 190–20 DOI: 10.1007/s10147-006-0583-4 PMID: 16850125
24. Sharma SV et al (2007) Epidermal growth factor receptor mutations in lung cancer Nat Rev Cancer 7(3) 169–81 DOI: 10.1038/nrc2088 PMID: 17318210
25. Murray S et al (2008) Somatic mutations of the tyrosine kinase domain of the epidermal growth factor receptor and tyrosine kinase inhibitor response to TKIs in non-small cell lung cancer: an analytical database J Thorac Oncol 3(8) 832–9 DOI: 10.1097/JTO.0b013e31818071f3 PMID: 18670300
26. Wu JY et al (2011) Effectiveness of tyrosine kinase inhibitors on «uncommon» epidermal growth factor receptor mutations of unknown clinical significance in non-small cell lung cancer Clin Cancer Res 17(11) 3812–21 DOI: 10.1158/1078-0432.CCR-10-3408 PMID: 21531810
27. De Pas T et al (2011) Activity of epidermal growth factor receptor-tyrosine kinase inhibitors in patients with non-small cell lung cancer harbouring rare epidermal growth factor receptor mutations J Thorac Oncol 6(11) 1895–901 DOI: 10.1097/JTO.0b013e318227e8c6 PMID: 21841502
28. Yang JC et al (2013) Activity of afatinib in uncommon epidermal growth factor receptor (EGFR) mutations: findings from three trials of afatinib in EGFR mutation-positive lung cancer Oral presentation at: World Congress on Lung Cancer
29. Chiu CH et al (2015) Epidermal growth factor receptor tyrosine kinase inhibitor treatment response in advanced lung adenocarcinomas with G719X/L861Q/S768I mutations J Thorac Oncol 10(5) 793–9 DOI: 10.1097/JTO.0000000000000504 PMID: 25668120
30. Yang JC et al (2013) Symptom control and quality of life in LUX-Lung 3: a phase III study of afatinib or cisplatin/pemetrexed in patients with advanced lung adenocarcinoma with EGFR mutations J Clin Oncol 31(27) 3342–50 DOI: 10.1200/JCO.2012.46.1764 PMID: 23816967
31. Yang JC-H et al (2014) Overall survival (OS) in patients (pts) with advanced non-small cell lung cancer (NSCLC) harboring common (Del19/L858R) epidermal growth factor receptor mutations (EGFR mut): pooled analysis of two large open-label phase III studies (LUX-Lung 3 [LL3] and LUX-Lung 6 [LL6]) comparing afatinib with chemotherapy (CT) J Clin Oncol 32(Suppl):abstr 8004
32. Jackman D et al (2010) Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer J Clin Oncol 28(2) 357–60 DOI: 10.1200/JCO.2009.24.7049
33. Wu JY et al (2008) Lung cancer with epidermal growth factor receptor exon 20 mutations is associated with poor gefitinib gefitinib tretment response Clin Cancer Res 14(15) 4877–82 DOI: 10.1158/1078-0432.CCR-07-5123 PMID: 18676761
34. Su KY et al (2012) Pretreatment epidermal growth factor receptor (EGFR) T790M mutation predicts shorter EGFR tyrosine kinase inhibitor response duration in patients with non-small-cell lung cancer J Clin Oncol 30(4) 433–40 DOI: 10.1200/JCO.2011.38.3224 PMID: 22215752
35. Costa C et al (2014) The impact of EGFR T790M mutations and BIM mRNA expression on outcome in patients with EGFR-mutant NSCLC treated with erlotinib or chemotherapy in the randomized phase III EURTAC trial Clin Cancer Res 20(7) 2001–10 DOI: 10.1158/1078-0432.CCR-13-2233 PMID: 24493829
36. Yamamoto C et al (2010) Loss of PTEN expression by blocking nuclear translocation of EGR1 in gefitinib-resistant lung cancer cells harboring epidermal growth factor receptor-activating mutations Cancer Res 70(21) 8715–25 DOI: 10.1158/0008-5472.CAN-10-0043 PMID: 20959484
37. Sos ML et al (2009) PTEN loss contributes to erlotinib resistance in EGFR-mutant lung cancer by activation of Akt and EGFR Cancer Res 69(8) 3256–61 DOI: 10.1158/0008-5472.CAN-08-4055 PMID: 19351834 PMCID: 2849653
38. Wang L et al (2014) PIK3CA mutations frequently coexist with EGFR/KRAS mutations in non-small cell lung cancer and suggest poor prognosis in EGFR/KRAS wildtype subgroup PLoS One 9(2) e88291 DOI: 10.1371/journal.pone.0088291 PMID: 24533074 PMCID: 3922761
39. Engelman JA et al (2006) Allelic dilution obscures detection of a biologically significant resistance mutation in EGFR-amplified lung cancer J Clin Invest 116(10) 2695–706 DOI: 10.1172/JCI28656 PMID: 16906227 PMCID: 1570180
40. Ng KP et al (2012) A common BIM deletion polymorphism mediates intrinsic resistance and inferior responses to tyrosine kinase inhibitors in cancer Nat Med 18(4) 521–8 DOI: 10.1038/nm.2713 PMID: 22426421
41. Costa C et al (2014) The impact of EGFR T790M mutations and BIM mRNA expression on outcome in patients with EGFR-mutant NSCLC treated with erlotinib or chemotherapy in the randomized phase III EURTAC trial Clin Cancer Res 20(7) 2001–10 DOI: 10.1158/1078-0432.CCR-13-2233 PMID: 24493829
42. Ohashi K et al (2013) Epidermal growth factor receptor tyrosine kinase inhibitor-resistant disease J Clin Oncol 31(8) 1070–80 DOI: 10.1200/JCO.2012.43.3912 PMID: 23401451 PMCID: 3589701
43. Yun CH et al (2008) The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP Proc Natl Acad Sci USA 105(6) 2070–5 DOI: 10.1073/pnas.0709662105 PMID: 18227510 PMCID: 2538882
44. Maheswaran S et al (2008) Detection of mutations in EGFR in circulating lung-cancer cells N Engl J Med 359(4) 366–77 DOI: 10.1056/NEJMoa0800668 PMID: 18596266 PMCID: 3551471
45. Balak MN et al (2006) Novel D761Y and common secondary T790M mutations in epidermal growth factor receptor-mutant lung adenocarcinomas with acquired resistance to kinase inhibitors Clin Cancer Res 12(21) 6494–501 DOI: 10.1158/1078-0432.CCR-06-1570 PMID: 17085664
46. Bean J et al (2008) Acquired resistance to epidermal growth factor receptor kinase inhibitors associated with a novel T854A mutation in a patient with EGFR-mutant lung adenocarcinoma Clin Cancer Res 14(22) 7519–25 DOI: 10.1158/1078-0432.CCR-08-0151 PMID: 19010870 PMCID: 2596620
47. Nguyen KS, Kobayashi S and Costa DB (2009) Acquired Resistance to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Non–Small-Cell Lung Cancers Dependent on the Epidermal Growth Factor Receptor Pathway Clin Lung Cancer 10(4) 281–9 DOI: 10.3816/CLC.2009.n.039 PMID: 19632948 PMCID: 2758558
48. Engelman JA et al (2007) MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling Science 316(5827) 1039–43 DOI: 10.1126/science.1141478 PMID: 17463250
49. Yano S et al (2008) Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor-activating mutations Cancer Res 68(22) 9479–87 DOI: 10.1158/0008-5472.CAN-08-1643 PMID: 19010923
50. Yano S et al (2011) Hepatocyte growth factor expression in EGFR mutant lung cancer with intrinsic and acquired resistance to tyrosine kinase inhibitors in a Japanese cohort J Thorac Oncol 6(12) 2011–7 DOI: 10.1097/JTO.0b013e31823ab0dd PMID: 22052230
51. Turke AB et al (2010) Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC Cancer Cell 17(1) 77–88 DOI: 10.1016/j.ccr.2009.11.022 PMID: 20129249 PMCID: 2980857
52. Sequist LV et al (2011) Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors Sci Transl Med 3(75) 75ra26 PMID: 21430269 PMCID: 3132801
53. Yu HA et al (2013) Analysis of tumour specimens at the time of acquired resistance to EGFR TKI therapy in 155 patients with EGFR-mutant lung cancers Clin Cancer Res 19(8) 2240–7 DOI: 10.1158/1078-0432.CCR-12-2246 PMID: 23470965 PMCID: 3630270
54. Thiery JP and Sleeman JP (2006) Complex networks orchestrate epithelial-mesenchymal transitions Nat Rev Mol Cell Biol 7(2) 131–42 DOI: 10.1038/nrm1835 PMID: 16493418
55. Suda K et al (2011) Epithelial to mesenchymal transition in an epidermal growth factor receptor mutant lung cancer cell line with acquired resistance to erlotinib J Thorac Oncol 6(7) 1152–61 DOI: 10.1097/JTO.0b013e318216ee52 PMID: 21597390
56. Zavadil J and Bottinger EP (2005) TGF-β and epithelial-to-mesenchymal transitions Oncogene 24(37) 5764–74 DOI: 10.1038/sj.onc.1208927 PMID: 16123809
57. Xie M et al (2012) Activation of Notch-1 enhances epithelial-mesenchymal transition in gefitinib acquired resistant lung cancer cells J Cell Biochem 113(5) 1501–13
58. Zhang Z et al (2012) Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer Nat Genet 44(8) 852–60 DOI: 10.1038/ng.2330 PMID: 22751098 PMCID: 3408577
59. Yu HA et al (2013) Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers Clin Cancer Res 19(8) 2240–7 DOI: 10.1158/1078-0432.CCR-12-2246 PMID: 23470965 PMCID: 3630270
60. Lin PY, Yu SL and Yang PC (2010) MicroRNA in lung cancer Br J Cancer 103(8) 1144–8 DOI: 10.1038/sj.bjc.6605901 PMID: 20859290 PMCID: 2967070
61. Krol J, Loedige I and Filipowicz W (2010) The widespread regulation of microRNA biogenesis, function and decay Nat Rev Genet 11(9) 597–610 PMID: 20661255
62. Qi J and Mu D (2012) MicroRNAs and lung cancers: from pathogenesis to clinical implications Front Med 6(2) 134–55 DOI: 10.1007/s11684-012-0188-4 PMID: 22528868 PMCID: 3725603
63. Grignani F et al (2014) Correlation of circulating miRNA levels with progression-free survival (PFS) and overall survival (OS) in early-stage lung adenocarcinoma J Clin Oncol 32 (suppl 5; abstr 11099)
64. Pan X, Wang ZX and Wang R (2010) MicroRNA-21 A novel therapeutic target in human cancer Cancer Biol Ther 10(12) 1224–32 DOI: 10.4161/cbt.10.12.14252 PMID: 21139417
65. Frezzetti D et al (2011) Upregulationof miR-21 by Ras in vivo and its role in tumor growth Oncogene 30(3) 275–86 DOI: 10.1038/onc.2010.416
66. Seike M et al (2009) MiR-21 is an EGFR-regulated antiapoptotic factor in lung cancer in never-smokers Proc Natl Acad Sci U S A 106(29) 12085–90 DOI: 10.1073/pnas.0905234106 PMID: 19597153 PMCID: 2715493
67. Li B et al (2014) MiR-21 overexpression is associated with acquired resistance of EGFR-TKI in non-small cell lung cancer Lung Cancer 83(2) 146–53 DOI: 10.1016/j.lungcan.2013.11.003
68. Shen H et al (2014) Alteration in Mir-21/PTEN Expression Modulates Gefitinib Resistance in Non-Small Cell Lung Cancer PLoS One 9(7) e103305 DOI: 10.1371/journal.pone.0103305 PMID: 25058005 PMCID: 4110008
69. Wang YS et al (2012) MicroRNA-214 regulates the acquired resistance to gefitinib via the PTEN/AKT pathway in EGFR-mutant cell lines Asian Pac J Cancer Prev 13(1) 255–60 DOI: 10.7314/APJCP.2012.13.1.255 PMID: 22502680
70. Cao M et al (2012) MiR-23a regulates TGF-beta-induced epithelial-mesenchymal transition by targeting E-cadherin in lung cancer cells Int J Oncol 41(3) 869–75 PMID: 22752005 PMCID: 3582905
71. Izumchenko E et al (2014) The TGFβ-miR200-MIG6 pathway orchestrates the EMT-associated kinase switch that induces resistance to EGFR inhibitors Cancer Res 74(14) 3995–4005 DOI: 10.1158/0008-5472.CAN-14-0110 PMID: 24830724 PMCID: 4122100
72. Kitamura K et al (2014) MiR-134/487b/655 cluster regulates TGF-β-induced epithelial-mesenchymal transition and drug resistance to gefitinib by targeting MAGI2 in lung adenocarcinoma cells Mol Cancer Ther 13(2) 444–53 DOI: 10.1158/1535-7163.MCT-13-0448
73. Cragg MS et al (2007) Gefitinib-induced killing of NSCLC cell lines expressing mutant EGFR requires BIM and can be enhanced by BH3 mimetics PLoS Med 4(10) 1681–9 DOI: 10.1371/journal.pmed.0040316 PMID: 17973573 PMCID: 2043013
74. Garofalo M et al (2011) EGFR and MET receptor tyrosine kinase-altered microRNA expression induces tumorigenesis and gefitinib resistance in lung cancers Nat Med 18(1) 74–82 PMID: 22157681 PMCID: 3467100
75. Zhou YM, Liu J and Sun W (2014) MiR-130a Overcomes Gefitinib Resistance by Targeting Met in Non-Small Cell Lung Cancer Cell Lines Asian Pac J Cancer Prev 15(3) 1391–6 DOI: 10.7314/APJCP.2014.15.3.1391 PMID: 24606471
76. Acunzo M et al (2012) miR-130a targets MET and induces TRAIL-sensitivity in NSCLC by downregulating miR-221 and 222 Oncogene 31(5) 634–42
77. Garofalo M et al (2009) miR-221&222 regulate TRAIL resistance and enhance tumorigenicity through PTEN and TIMP3 downregulation Cancer Cell 16(6) 498–509 DOI: 10.1016/j.ccr.2009.10.014 PMID: 19962668 PMCID: 2796583
78. Zhou JY et al (2014) MicroRNA-34a overcomes HGF-mediated gefitinib resistance in EGFR mutant lung cancer cells partly by targeting MET Cancer Lett 351(2) 265–71 DOI: 10.1016/j.canlet.2014.06.010 PMID: 24983493
79. Gomez GG, Wykosky J and Zanca C (2013) Therapeutic resistance in cancer: microRNA regulation of EGFR signaling networks Cancer Biol Med 10(4) 192–205 PMID: 24349829 PMCID: 3860350
80. Raver-Shapira N et al (2007) Transcriptional activation of miR-34a contributes to p53-mediated apoptosis Mol Cell 26(5) 731–43 DOI: 10.1016/j.molcel.2007.05.017 PMID: 17540598
81. Kasinski AL and Slack FJ (2012) miRNA-34 prevents cancer initiation and progression in a therapeutically resistant K-ras and p53- induced mouse model of lung adenocarcinoma Cancer Res 72(21) 5576–87 DOI: 10.1158/0008-5472.CAN-12-2001 PMID: 22964582 PMCID: 3488137
82. Zhao J, Kelnar K and Bader AG (2014) In-depth analysis shows synergy between erlotinib and miR-34a PLoS One 9(2) e89105 DOI: 10.1371/journal.pone.0089105 PMID: 24551227 PMCID: 3925231
83. Mudduluru G et al (2011) Regulation of Axl receptor tyrosine kinase expression by miR-34a and miR-199a/b in solid cancer Oncogene 30(25) 2888–99 DOI: 10.1038/onc.2011.13 PMID: 21317930
84. Kaller M et al (2011) Genome-wide characterization of miR-34a induced changes in protein and mRNA expression by a combined pulsed SILAC and microarray analysis Mol Cell Proteomics 10(8) M111.010462 DOI: 10.1074/mcp.M111.010462 PMID: 21566225 PMCID: 3149097
85. Gao Y et al (2014) miR-138-5p reverses gefitinib resistance in non-small cell lung cancer cells via negatively regulating G protein-coupled receptor 124 Biochem Biophys Res Commun 446(1) 179–86 DOI: 10.1016/j.bbrc.2014.02.073 PMID: 24582749
86. Rai K et al (2011) Liposomal delivery of MicroRNA-7-expressing plasmid overcomes epidermal growth factor receptor tyrosine kinase inhibitor-resistance in lung cancer cells Mol Cancer Ther 10(9) 1720–7 DOI: 10.1158/1535-7163.MCT-11-0220 PMID: 21712475
87. Lee CG et al (2014) MicroRNA-147 induces a mesenchymal-to-epithelial transition (MET) and reverses EGFR inhibitor resistance PLoS One 9(1) e84597 DOI: 10.1371/journal.pone.0084597 PMID: 24454732 PMCID: 3893127
88. Chen JC et al (2014) Suppression of dicer increases sensitivity to gefitinib in human lung cancer cells Ann Surg Oncol 21(Suppl 4) 555-63 DOI: 10.1245/s10434-014-3673-y
89. Ricciuti B et al (2014) Non-coding RNAs in lung cancer Oncoscience 1(11) 674–705