Dihydropyrimidine dehydrogenase (DPD) polymorphisms knocking on the door
Mauro Daniel Spina Donadioa, Dirce Maria Carraro, Giovana Tardin Torrezan and Celso Abdon Lopes de Mello
AC Camargo Cancer Center, R Prof Antonio Prudente 211, Liberdade, São Paulo, SP 01509010, Brazil
ahttps://orcid.org/0000-0002-4705-4802
Abstract
Identifying polymorphisms in the dihydropyrimidine dehydrogenase (DPYD) genes is gaining importance as predictors of fluoropyrimidine-associated toxicity. The recommendation of dose adjustment for chemotherapy guided by the presence of polymorphisms of the DPYD gene can potentially improve treatment safety for a large number of patients, saving lives, avoiding complications and reducing health care costs. This article discusses how personalisation of fluoropyrimidine treatment based on the identification of DPYD variants can mitigate toxicities and be cost effective.
Keywords: fluoropyrimidine, polymorphisms, dihydropyrimidine dehydrogenase
Correspondence to: Mauro Daniel Spina Donadio
Email: mauro.donadio@accamargo.org.br
Published: 17/01/2022
Received: 17/08/2021
Publication costs for this article were supported by ecancer (UK Charity number 1176307).
Copyright: © the authors; licensee ecancermedicalscience. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Background
Fluoropyrimidines are one of the most widely used chemotherapy drugs against solid cancers, either as monotherapy or in combination therapy, and more than 2 million cancer patients are exposed annually to this drug, which includes 5-fluorouracil (5-FU) and its oral pro-drugs capecitabine and tegafur [1]. Like all other chemotherapy drugs, fluoropyrimidines also cause toxicities. Adverse drug reactions are a major clinical problem during chemotherapy treatment and often require dose reduction and even treatment interruption. Unfortunately, 10%–30% of patients treated with fluoropyrimidines experience severe or potentially fatal treatment-related toxicity and in 0.5%–1% of these patients the toxicity is lethal [2, 3]. The main adverse events caused by fluoropyrimidines are haematological (leukopenia including febrile neutropenia, anaemia and thrombocytopenia), gastrointestinal (mucositis, stomatitis, diarrhoea, nausea and vomiting) and dermatological (hand-foot syndrome, hair loss and dry skin) but most of these events are mild, reversible and controlled with support measures [4].
In patients with certain enzyme deficiencies that act on the fluoropyrimidine metabolism, however, the use of these chemotherapeutic agents can lead to life-threatening complications, including severe nausea, vomiting and diarrhoea with volume depletion, extensive skin and mucositis changes, pancytopenia with risk of bleeding and infection, cardiotoxicity and neurological abnormalities such as cerebellar ataxia, cognitive dysfunction and altered level of consciousness [5–13]. In these cases, toxicity can occur early during the first treatment cycle, reinforcing the importance of detecting these enzyme deficiencies before the start of therapy, so that personalised dose adjustments of fluoropyrimidine, or even alternative drugs, can be prescribed [14].
The fluoropyrimidine toxicity involves a complex and multi-step mechanism responsible for the drug and its products metabolism and excretion. One of the main steps in the cascade of 5-FU metabolism involves the dihydropyrimidine dehydrogenase (DPD) enzyme, coded by the DPYD gene. The genetic factor is the main factor responsible for this enzyme activity and polymorphisms can eventually modify drug metabolism, resulting in drug accumulation and toxicity. Interindividual genetic variation in certain genes is responsible for a significant proportion of adverse reactions and can identify biomarkers that are predictive of the risk of toxicity associated with fluoropyrimidine [15, 16]. Identifying these variants, then, is a relevant effort because it has the potential to greatly improve the safety of a large number of patients.
We conducted a critical review on the mechanisms of fluoropyrimidine toxicity focusing on new molecular findings and recommendations. Moreover, we explored the burden of DPD testing in a developing country such as Brazil.
Fluoropyrimidines and metabolism pathways
The main fluoropyrimidine is 5-FU, an antimetabolite chemotherapeutic agent that was developed in 1957 by Heidelberger [17]. This drug is widely used in many neoplasms and is a cornerstone treatment for gastrointestinal malignancies. 5-FU is a prodrug that requires intracellular conversion to cytotoxic metabolites with antitumour effects. Of the entire dose administered, the majority is degraded by the catabolic pathway (about 80%), another part is directly excreted in the urine and only 1%–3% of the 5-FU is anabolised to cytotoxic metabolites [18–21].
In the anabolic pathway, 5-FU is metabolised in tissues to 5-fluoro-2′-deoxyuridine and then to 5-fluoro-2′-deoxyuridine-5ine-monophosphate, the active metabolite of the drug. The active metabolite inhibits the enzyme thymidylate synthase, resulting in inhibition of DNA synthesis and repair, inducing cell apoptosis. In addition, the toxic effects resulting from the partial incorporation of 5-FU and its metabolites in DNA and RNA contribute to the mechanism of action of the drug [22, 23]. If there is reduced activity of the enzymes involved in the catabolism of 5-FU, the result can be a substantial increase in the drug’s half-life and, therefore, an increased risk of severe toxicity [18–21].
The second most used fluoropyrimidine is Capecitabine that is metabolised to 5-FU in three consecutive steps, first metabolised to 5′-deoxy-5-fluorocytidine by carboxylesterase, which is subsequently converted to 5′-deoxy-5-fluorouridine by cytidine deaminase and finally to 5-FU by thymidine phosphorylase enzyme. Tegafur, in turn, is metabolised to 5-FU and to the less cytotoxic metabolites 3-hydroxytegafur, 4-hydroxytegafur and dihydrotegafur by Cytochrome P450 2A6 (CYP2A6) [22, 23].
Genetic variants and their impact on fluoropyrimidines
Genetic variants in the genes coding the metabolic pathway enzymes can alter the metabolism of 5-FU and are clinically significant predictors of fluoropyrimidine toxicity: genetic polymorphisms of the TYMS gene (responsible for the enzyme thymidylate synthase) and the enzyme methylenetetrahydrofolate reductase gene are described, although rare. In addition, the variation in cytidine deaminase (CDA) expression was associated with polymorphism in the CDA promoter region, with an impact on gemcitabine and capecitabine metabolism [15].
The most well-known genetic variant in this scenario is the deficiency of the 5-FU metabolic enzyme, DPD. In 39%–61% of patients with severe toxicity to chemotherapy, the reduced activity in the peripheral blood mononuclear cells of this enzyme has been found [16].
Variants in DPYD, the gene that encodes DPD, are gaining importance as predictors of fluoropyrimidine-associated toxicity because the serum tests that detect them are increasingly available and, based on them, dose adaptation is now recommended by some guidelines, such as the Clinical Pharmacogenetics Implementation Consortium [24], Dutch Pharmacogenetics Working Group [25], already endorsed by the European Association of Clinical and Therapeutic Pharmacology and the European Association of Hospital Pharmacists [25, 26]. In 2020, the European Medicines Agency recommended preventive testing for DPYD variants before starting cancer treatment with 5-FU, capecitabine and tegafur [27]. This recommendation, however, has not yet been endorsed by the Food and Drug Administration (FDA), European Society of Medical Oncology or the National Comprehensive Cancer Network.
In the catabolic pathway, DPD is the first enzyme that acts by converting 5-FU into dihydrouracil (FUH2) and, although the enzyme has been shown to be present in several tissues, it is believed that the liver is the main organ responsible for 5-FU catabolism. After this conversion, the FUH2 is subsequently metabolised to its final metabolite 5-fluoro-β-alanine, which is excreted in the urine[4]. Next to converting 5-FU, the DPD enzyme also converts its endogenous substrate uracil (U) into dihydrouracil (DHU). The pretreatment ratio of serum DHU/U concentrations was investigated as a phenotypic measure of DPD activity. However, the clinical applicability of the DHU/U ratio has been limited by the lack of robust evidence on clinical validity [4, 16].
There are some possible methods to evaluate DPD function and verifying DPD activity: measuring DPD enzyme activity in peripheral blood mononuclear cells; the 2-¹³C-uracil breath test (where ¹³C02 is measured, which is a product of the degradation of 2-¹³C-uracil by DPD and other enzymes involved in the catabolic route of pyrimidines), the quantification of the DHU/U ratio in plasma and measuring the metabolism of a single dose of uracil [23]. However, all DPD phenotyping tests have their limitations and measuring DPD activity in advance on a routine basis is technically and logistically challenging, laborious and expensive [23].
DPYD is a highly polymorphic gene, located on chromosome 1p22, with a single copy of 950 kb that covers 23 exons and more than 7,600 genetic variants have been recorded. Although the majority of these variants are intronic variants that can be considered silent, part of this genetic variation is considered responsible for the great variability in DPD activity that is observed in the general population [4, 23].
In fact, several of the investigated variants have been reported to be associated with reduced enzyme activity and have been proposed as potentially associated with severe 5-FU toxicity, but of these variants, only four were consistently associated with a marked decrease in DPD activity and increased toxicity of fluoropyrimidine, with ≥3 grade toxicity according to the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) related to 5-FU in case–control studies [4]. These variants include DPYD*2A single nucleotide polymorphism (SNP) (c.1905+ 1G>A), DPYD*13 SNP (c.1679T>G), SNP c.2846A>T and a collection of SNPs called HapB3 (a new haplotype – hapB3, composed of some variants, such as: c.483+18G>A; c.680+139G>A; c.959-51T>G; c.1236G>A and the likely causal c.1129-5923C>G intronic variant) [16, 28–30].
The initial screening for the most well-known variant, c.1905+1G>A (previously called IVS14+1G>A or DPD*2A), and dose individualisation in patients with this allele has already been shown to improve treatment safety, avoiding fluoropyrimidine associated severe and potentially fatal toxicity [16, 24]. This variant is the most studied in the context of 5-FU toxicity and the first studies suggested that it would be responsible for up to 29% of all toxicities of grade ≥ 3 but, despite recognising that patients with this variant are at increased risk of severe 5-FU toxicity, the proportion of toxicity cases that could be explained by its presence varies widely. In the largest cohort of more than 680 patients treated with 5-FU monotherapy, 5.5% of 5-FU toxicity cases were explained by c.1905+1G>A [31].
Current data suggest that these variants combined are an important contributing factor for the occurrence of adverse events, accounting for at least 20% of the observed cases of severe toxicities related to 5-FU [4].
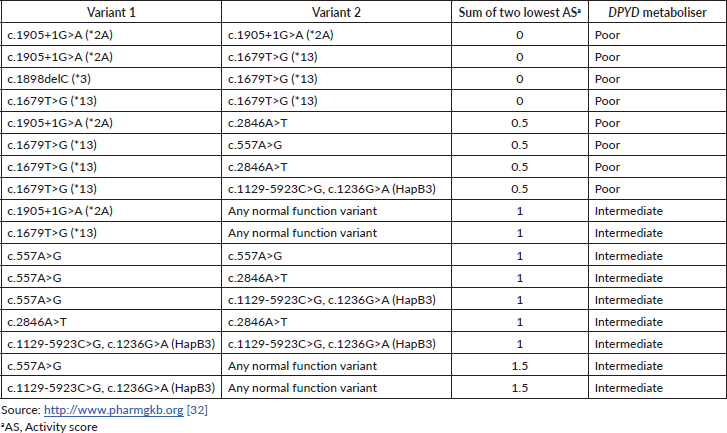
The DPD phenotype is assigned using a gene activity score (AS) based on the DPYD allele functionality (as shown in Table 1) and calculated as the sum of the two DPYD variants with the lowest variant activity value [32]. Table 2 contains the main examples of diplotypes present in available commercial tests with the respective AS and their impact on the DPD metaboliser activity.
Table 1. Activity value and functional status of strong evidence-based DPYD alleles.
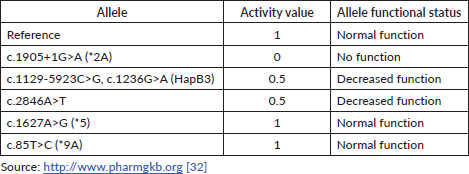
Table 2. Examples of diplotypes with respective AS and DPYD metaboliser predictors.
The difficulties of analysing individual variants
In addition to a relative consistency between studies in the proportion of toxicity cases that can be explained by the sum of multiple variants of DPYD, given a comprehensive genetic screening of the gene, the importance of individual variants was more variable between studies. There are several potential explanations for these variable results in relation to relatively rare individual variants, well discussed by Amstutz et al [4]:
• Population frequency differences: a small allele frequency difference for a rare deleterious allele in different populations can lead to large carrier frequency differences and accentuate their relative importance for 5-FU toxicity.
• Sampling effects: because the DPYD variants have such low frequencies, it would be necessary to evaluate a very high number of toxicity events to arrive at a reliable estimate of the importance of a specific variant for a serious adverse event. Still, this can vary with each individual variant. To mitigate this, the ideal approach would be to combine information from multiple variants with comprehensive genetic screening.
• Therapy-related heterogeneity: a considerable source of inconsistencies in the results of different studies related to 5-FU toxicity in DPD deficiency is treatment-related heterogeneity as the functional relevance of DPYD variation may vary between different treatment regimens and doses of 5-FU. In addition, there is an overlap of toxicities between chemotherapeutic agents, which can increase the risk of adverse effects, as well as drug interactions that directly affect the metabolism of 5-FU, modifying the risk profile for DPYD variants. Another aspect is the sequencing of therapy on DPD function, for example, prior use of gemcitabine can induce liver tissue damage and severe toxicity with capecitabine even in the absence of DPD dysfunction.
• Heterogeneity in toxicity assessment: another source of inconsistency in the results of different studies related to 5-FU toxicity in DPD deficiency is the form of assessment of 5-FU toxicity. In addition to using different grading criteria for adverse effects, some studies evaluated toxicities at different times during treatments, that is, severe toxicity was not always characterised according to NCI CTCAE grade 3 to 5 in early chemotherapy cycles.
Table 3. The allelic frequency of the four main DPYD variants according to the ABraOM repository.
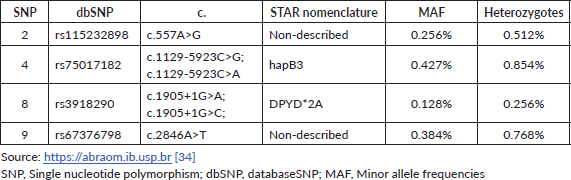
The frequency of the various DPYD variants and the associated phenotypes appears to vary significantly between ethnic groups. Considering all four main variants combined, 5%–7% of the white population has a partial deficiency and 0.1%–0.2% has a complete deficiency of the DPD enzyme. On the other hand, about 8% of the African American population has partial DPD deficiency [23, 33]. The Brazilian population is constituted by nearly 500 years of admixture between Africans, Europeans, Native Americans and Japanese enabling peculiar genetic combinations. The allelic frequency of the four main variants according to the Online Archive of Brazilian Mutations (ABraOM) repository, which contains genomic variants identified by whole-exome and whole-genome sequencing from 1.171 unrelated elderly healthy individuals from São Paulo-Brazil, is shown in Table 3 [34]. As Brazil is a large country with great ethnic diversity, DPYD allele frequencies are not homogeneous across its subpopulations and studies with specific subpopulations show different allele prevalence. In example, data from 146 individuals from three Amazonian Amerindian populations showed minor allele frequencies of 1% and 2% for DPYD*2A and DPYD*13, and in healthy Brazilians of predominantly African ancestry or self-reported as black the c.557A>G variant was detected at a frequency of 2.6% [35, 36]. For further analysis and discussions in the text, data from the ABraOM repository will be used as a parameter.
Dose adjustment recommendation guidelines
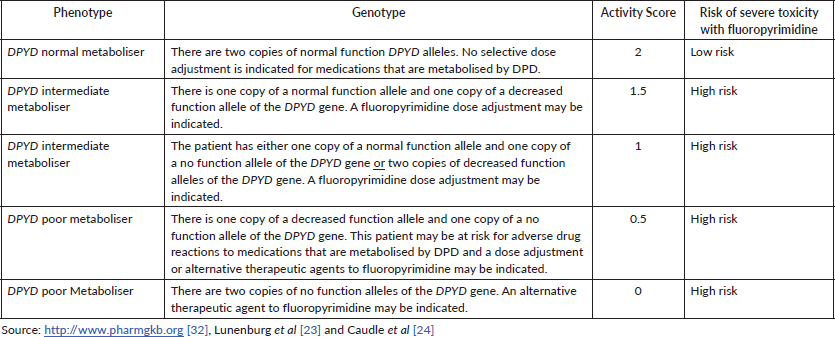
Patients with low DPD activity are expected to have an increased risk of developing severe or even lethal toxicity when treated with standard doses of 5-FU or capecitabine [23]. Predicted DPD activity can be expressed as the DPYD gene AS, which ranges from 0 (none or practically no DPD enzyme activity) to 2 (normal DPD enzyme activity due to homozygosity for fully functional alleles, both attributed to an AS 1). The gene AS is a sum of the two activities of the protein isoforms expressed in both alleles [23, 33]. Carriers of two no function variants (AS 0) or one decreased function variant (AS 0.5) are classified as DPYD poor metabolisers; carriers of two decreased function variants or carriers of only one no function variant (AS 1) or carriers of only one decreased function variant (AS 1.5) are considered DPYD intermediate metabolisers, and those with only normal function variants are classified as DPYD normal metabolisers (AS 2). Each decreased or no function variant is considered to be on a different gene copy and patients may carry multiple normal function variants. As an individual only carries a maximum of two fully functional DPYD copies, common normal function variants may be located on the same gene copy as other normal function variants or decreased or no function variants [32].
The guidelines that address the topic, in summary, suggest that individuals with a gene AS of 0 or 0.5 are recommended to avoid 5-FU, capecitabine or tegafur; individuals with a genetic AS of 1 or 1.5 are recommended to initiate therapy with at least 50% of the standard dose of 5-FU or capecitabine but avoid tegafur. A gene AS of 2 (reference value) does not result in a recommendation for dose adjustment for 5-FU, capecitabine or tegafur [23, 33].
Therefore, high-risk patients with DPYD risk alleles could receive modified doses of 5-FU or monotherapy as an alternative treatment option with a potentially increased survival benefit compared to a complete discontinuation of 5-FU therapy [4]. Table 4 shows the correlation between genotype, phenotype, DPD AS and respective risk of severe toxicity associated with fluoropyrimidine.
Although the combination of 5-FU with newer cytotoxic agents, for example, the third-generation platinum derivative oxaliplatin or the topoisomerase I inhibitor irinotecan, or targeted therapies such as bevacizumab, cetuximab or panitumumab, resulted in rates of response significantly improved, the effectiveness of the same agents without the 5-FU combination was limited [37].
Table 4. Assignment of likely DPD phenotype based on genotype and respective toxicity risk.
For homozygous patients carriers of two identical non-functional alleles and compound heterozygous patients carriers of two different non-functional alleles, it is necessary to use alternative agents. The quinazoline folate analogue raltitrexed, which is a thymidylate synthase inhibitor, may be a useful substitute for 5-FU in patients with DPD deficiency, but it is not widely available [38]. Other reported strategies include use of trifluridine-tipiracil (TAS-102) instead of fluoropyrimidine or fluoropyrimidine micro-dosing [39, 40].
Supportive treatment after severe toxicity associated with DPD deficiency
Most cases of DPD deficiency are diagnosed only after a severe reaction to 5-FU. The management of these patients should include aggressive haemodynamic support, parenteral nutrition, antibiotics, granulocyte colony-stimulating factors (G-CSF) and, when available, uridine triacetate (UT). UT is a specific pharmacological antidote for fluoropyrimidines, an orally administered uridine prodrug approved by the FDA for emergency use after an overdose of 5-FU or capecitabine. It must be administered within 96 hours after the end of the administration of these chemotherapeutic agents. The recommended dose is 10 g orally every 6 hours, making a total of 20 doses. Despite its approval, UT has a high cost and is not commercially available [41, 42].
This supportive treatment for patients with DPD deficiency presenting severe 5-FU toxicity is based only on case reports and the ideal management still lacks evidence. The use and timing of G-CSF, for example, needs to be better assessed and discussed. Studies of preclinical models that do not involve DPD-related toxicity suggest that G-CSF should not be used early [43]. In individuals with higher and sustained serum levels of cytotoxic agents, as in DPD deficiency, the use of early G-CSF may actually be counterproductive and, to assess the best time of use, it may be necessary to dose the serum level of uracil, for example.
Cost-effectiveness of routine screening
The costs of prospectively carrying out the DPYD gene polymorphism tests appear to be effective. In fact, they would save a greater expenditure on supportive care although it is not possible to price preventable death.
A cost-effectiveness study by an Irish institution evaluated 134 patients who started chemotherapy with first-line fluoropyrimidine over 3 years. Thirty (23%) patients developed grade 3/4 toxicity. Of these, 17% revealed heterozygous DPYD deleterious alleles. The cost of hospitalisation for patients with a DPYD variant was € 232,061, while prospective testing of all 134 patients would have cost € 23,718. This study suggests that prospective tests would result in cost savings because the cost of hospital admission for severe chemotherapy-related toxicity is significantly higher than the cost of prospective DPYD testing for each patient starting fluoropyrimidine chemotherapy [44].
The discussion of cost effectiveness in this scenario is very pertinent since almost half a million patients in Brazil and more than 900,000 in South America have cancers that can be exposed to fluoropyrimidine at some point in the treatment of their disease (Table 5). Testing for DPD deficiency in these emerging countries is certainly a factor that impacts the cost of health care, often prohibitive. In Brazil, a single initiative in the public health system setting has been recently published [45]. However, considering the scenario of limited financial resources that these countries present, unfortunately we cannot envision the universal use of genetic tests in the short term.
It may be necessary to find a niche for patients at higher risk for having DPD deficiency or who would be more vulnerable to complications from chemo toxicities, such as morbid and elderly people, and prioritise testing for these groups. We also have to think about whether the group of patients who will receive higher doses of fluoropyrimidine should be prioritised. The risk of toxicity will always be greater with the use of regimens that use, for example, doses of infusional 5FU of 2,400 mg/m² plus 400 mg/m² in boluses, as in the use of 5-Fluorouracil, Leucovorin, Irinotecan and Oxaliplatin (FOLFIRINOX) for pancreatic cancer [46, 47], when compared with the Cyclophosphamide, Methotrexate and Fluorouracil (CMF) scheme that uses a dose of 600 mg/m², as in breast cancer [48]. There are no studies, however, that address the risk of toxicity by associating DPD deficiency and fluoropyrimidine dose. And this is just another unanswered question that will be increasingly asked in the care routine of oncology services.
As an example, Figure 1 shows the number of patients with colorectal cancer in Brazil according to stage, considering the prevalence according to Globocan, the sum of heterozygotes according to ABraOM, which implies a risk for up to 2.4% of the population, and the proportion per stage of the Surveillance, Epidemiology, and End Results database [52]. Considering colorectal cancer alone, from 48,015 patients with regional disease and 29,343 with distant metastasis, 1,152 and 704 patients would present a risk allele, respectively. It means that more than 1,800 colorectal cancer patients would be at risk of severe toxicity with the use of fluoropyrimidine in Brazil. Despite being an emerging country in which testing for pharmacogenetic variants can economically impact health care, the occurrence of serious toxicities in these patients would certainly have a greater economic impact, with great potential to lead to important morbidity and even death. Although the cost of these consequences cannot be precisely measured, they are potentially preventable if the right measures are taken after mutations are detected. Therefore, more research is needed to understand the cost effectiveness of DPYD screening in the setting of low- and middle-income countries such as Brazil.
Table 5. Incidence/prevalence of the most common cancers treated with fluoropyrimidines.
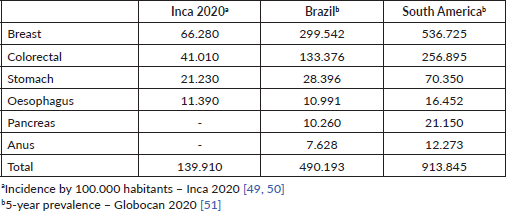
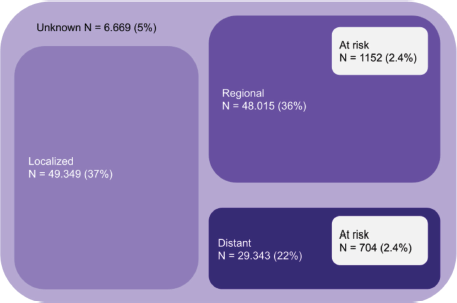
Figure 1. Absolute number of patients at risk considering only colorectal cancer in Brazil (patients at risk considering the sum of MAF (ABraOM)).
Conclusion
Comprehensive genetic testing of DPYD is needed in future studies involving the use of fluoropyrimidines. The recommendation of chemotherapy dose adjustment guided by the presence of DPYD polymorphisms can become mandatory in the near future due to the potential number of lives that can be saved, complications that can be avoided and costs that can be reduced worldwide. The DPYD genotyping and its applicability demand an urgent discussion regarding its standardisation, costs and indications. In the meantime, it is advisable to discuss with patients the rarity of these variants, but also their implications, considering the costs of pharmacogenetic tests. Despite the recognised relevance of these genomic tests, treatment with fluoropyrimidines should not be substantially modified until a definitive recommendation based on the medical oncology community is generated taking into account all aspects of this molecular approach including access, cost and accuracy. Studies are needed to try to discover and describe possible new deleterious variants of the DPYD gene for South American populations. Thus, investments in testing and treatment protocols or dose adjustment can be better targeted, optimising expenses in a scarce resources scenario.
Authors' contributions
All authors have made a significant contribution to this manuscript, have seen and approved the final manuscript and agree to its submission to the journal.
Conflicts of interest
None.
Funding
None to declare. This research did not receive any specific grant from funding agencies in the public, commercial or not-for-profit sectors.
Warnings
The opinions expressed in the report presented are those of the authors and do not necessarily represent the official position of the institution to which they belong.
References
1. Scrip’s Cancer Chemotherapy Report (2002) Scrip’s World Pharmaceutical News (London: PJB Publications Ltd)
2. Mikhail S, Sun J, and Marshall J, et al (2010) Safety of capecitabine: a review Expert Opin Drug 9 831–841 https://doi.org/10.1517/14740338.2010.511610
3. Lévy E, Piedbois P, and Buyse M, et al (1998) Toxicity of fluorouracil in patients with advanced colorectal cancer: effect of administration schedule and prognostic factors J Clin Oncol 16 3537–3541 https://doi.org/10.1200/JCO.1998.16.11.3537 PMID: 9817272
4. Amstutz U, Froehlich T, and Largiader C, et al (2011) Dihydropyrimidine dehydrogenase gene as a major predictor of severe 5-fluorouracil toxicity Pharmacogenomics 12 1321–1336 https://doi.org/10.2217/pgs.11.72 PMID: 21919607
5. Diasio R, Beavers T, and Carpenter J, et al (1988) Familial deficiency of dihydropyrimidine dehydrogenase. Biochemical basis for familial pyrimidinemia and severe 5-fluorouracil-induced toxicity J Clin Invest 81 47 https://doi.org/10.1172/JCI113308 PMID: 3335642 PMCID: 442471
6. Maring J, van Kuilenburg A, and Haasjes J, et al (2002) Reduced 5-FU clearance in a patient with low DPD activity due to heterozygosity for a mutant allele of the DPYD gene Br J Cancer 86 1028 https://doi.org/10.1038/sj.bjc.6600208 PMID: 11953843 PMCID: 2364178
7. Ezzeldin H, Johnson MR, and Okamoto Y, et al (2003) Denaturing high performance liquid chromatography analysis of the DPYD gene in patients with lethal 5-fluorouracil toxicity Clin Cancer Res 9 3021 PMID: 12912951
8. Harris B, Carpenter J, and Diasio R, et al (1991) Severe 5-fluorouracil toxicity secondary to dihydropyrimidine dehydrogenase deficiency. A potentially more common pharmacogenetic syndrome Cancer 68 499 https://doi.org/10.1002/1097-0142(19910801)68:3<499::AID-CNCR2820680309>3.0.CO;2-F PMID: 1648430
9. Takimoto C, Lu Z, and Zhang R, et al (1996) Severe neurotoxicity following 5-fluorouracil-based chemotherapy in a patient with dihydropyrimidine dehydrogenase deficiency Clin Cancer Res 2 477 PMID: 9816193
10. Ezzeldin H and Diasio R (2004) Dihydropyrimidine dehydrogenase deficiency, a pharmacogenetic syndrome associated with potentially life-threatening toxicity following 5-fluorouracil administration Clin Colorectal Cancer 4 181 https://doi.org/10.3816/CCC.2004.n.018 PMID: 15377401
11. Diasio R and Johnson M (1999) Dihydropyrimidine dehydrogenase: its role in 5-fluorouracil clinical toxicity and tumor resistance Clin Cancer Res 5 2672 PMID: 10537327
12. van Kuilenburg A, Meinsma R, and Zonnenberg B, et al (2003) Dihydropyrimidinase deficiency and severe 5-fluorouracil toxicity Clin Cancer Res 9 4363 PMID: 14555507
13. Yang C, Ciccolini J, and Blesius A, et al (2011) DPD-based adaptive dosing of 5-FU in patients with head and neck cancer: impact on treatment efficacy and toxicity Cancer Chemother Pharmacol 67 49 https://doi.org/10.1007/s00280-010-1282-4
14. Froehlich T, Amstutz U, and Aebi S, et al (2015) Clinical importance of risk variants in the dihydropyrimidine dehydrogenase gene for the prediction of early-onset fluoropyrimidine toxicity Int J Cancer 136 730–739
15. Loganayagam A, Arenas Hernandez M, and Corrigan A, et al (2013) Pharmacogenetic variants in the DPYD, TYMS, CDA and MTHFR genes are clinically significant predictors of fluoropyrimidine toxicity Br J Cancer 108 2505–2515 https://doi.org/10.1038/bjc.2013.262 PMID: 23736036 PMCID: 3694243
16. Meulendijks D, Henricks L, and Jacobs BAW, et al (2017) Pretreatment serum uracil concentration as a predictor of severe and fatal fluoropyrimidine-associated toxicity Br J Cancer 116 1415–1424 https://doi.org/10.1038/bjc.2017.94 PMID: 28427087 PMCID: 5520099
17. Heidelberger C, Chaudhuri NK, and Danneberg P, et al (1957) Fluorinated pyrimidines, a new class of tumour-inhibitory compounds Nature 179 663–666 https://doi.org/10.1038/179663a0 PMID: 13418758
18. Mattison L, Soong R, and Diasio R (2002) Implications of dihydropyrimidine dehydrogenase on 5-fluorouracil pharmacogenetics and pharmacogenomics Pharmacogenomics 3 485–492 https://doi.org/10.1517/14622416.3.4.485 PMID: 12164772
19. Diasio R and Harris B (1989) Clinical pharmacology of 5-fluorouracil Clin Pharmacokinet 16 215–237 https://doi.org/10.2165/00003088-198916040-00002 PMID: 2656050
20. Ezzeldin H and Diasio R (2004) Dihydropyrimidine dehydrogenase deficiency, a pharmacogenetic syndrome associated with potentially life-threatening toxicity following 5-fluorouracil administration Clin Col Cancer 4 181–189
21. van Kuilenburg A, Maring J, and Schalhorn A, et al (2008) Pharmacokinetics of 5-fluorouracil in patients heterozygous for the IVS14+1G > A mutation in the dihydropyrimidine dehydrogenase gene Nucleosides Nucleotides Nucleic Acids 27 692–698 https://doi.org/10.1080/15257770802145009 PMID: 18600527
22. Thorn C, Marsh S, and Carrillo M, et al (2011) PharmGKB summary: fluoropyrimidine pathways Pharmacogenet Genomics 21 237–242 https://doi.org/10.1097/FPC.0b013e32833c6107 PMCID: 3098754
23. Lunenburg C, van der Wouden C, and Nijenhuis M, et al (2020) Dutch Pharmacogenetics Working Group (DPWG) guideline for the gene–drug interaction of DPYD and fluoropyrimidines Eur J Hum Genet 28 508–517 https://doi.org/10.1038/s41431-019-0540-0
24. Caudle K, Thorn C, and Klein T, et al (2013) Clinical pharmacogenetics implementation consortium guidelines for dihydropyrimidine dehydrogenase genotype and fluoropyrimidine dosing Clin PharmacolTher 94 640–645
25. European Association for Clinical Pharmacology and Therapeutics (EACPT) [https://www.eacpt.eu/] Date accessed: 01/05/21
26. European Association of Hospital Pharmacists (EAHP) [https://www.eahp.eu/] Date accessed: 01/05/21
27. EMA provides new testing and treatment recommendations for fluorouracil capecitabine and tegafur [https://www.esmo.org/oncology-news/ema-provides-new-testing-and-treatment-recommendations-for-fluorouracil-capecitabine-and-tegafur] Date accessed: 01/05/21
28. Morel A, Boisdron-Celle M, and Fey L, et al (2006) Clinical relevance of different dihydropyrimidine dehydrogenase gene single nucleotide polymorphisms on 5-fluorouracil tolerance Mol Cancer Ther 5 2895 https://doi.org/10.1158/1535-7163.MCT-06-0327 PMID: 17121937
29. Offer S, Fossum C, and Wegner N, et al (2014) Comparative functional analysis of DPYD variants of potential clinical relevance to dihydropyrimidine dehydrogenase activity Cancer Res 74 2545 https://doi.org/10.1158/0008-5472.CAN-13-2482 PMID: 24648345 PMCID: 4012613
30. Amstutz U, Farese S, and Aebi S, et al (2009) Dihydropyrimidine dehydrogenase gene variation and severe 5-fluorouracil toxicity: a haplotype assessment Pharmacogenomics 10 931 https://doi.org/10.2217/pgs.09.28 PMID: 19530960
31. Schwab M, Zanger UM, and Marx C, et al (2008) Role of genetic and nongenetic factors for fluorouracil treatment-related severe toxicity: a prospective clinical trial by the German 5-FU Toxicity Study Group J Clin Oncol 26 2131–2138 https://doi.org/10.1200/JCO.2006.10.4182 PMID: 18299612
32. Gene-specific information tables for DPYD [https://www.pharmgkb.org/page/dpydRefMaterials] Date accessed: 01/05/21
33. Amstutz U, Henricks L, and Offer S, et al (2018) Clinical pharmacogenetics implementation consortium (CPIC) guideline for dihydropyrimidine dehydrogenase genotype and fluoropyrimidine dosing: 2017 update Clin Pharmacol Ther 103 210–216 https://doi.org/10.1002/cpt.911
34. ABraOM: Brazilian genomic variants [https://abraom.ib.usp.br] Date accessed: 01/08/21
35. Suarez-Kurtz G, Kovaleski G, and Elias A, et al (2020) Implementation of a pharmacogenomic program in a Brazilian public institution Pharmacogenomics 21 549–557 https://doi.org/10.2217/pgs-2020-0016 PMID: 32378980
36. Rodrigues J, Fernandes M, and Guerreiro J, et al (2019) Polymorphisms of ADME-related genes and their implications for drug safety and efficacy in Amazonian Amerindians Sci Rep 9 7201 https://doi.org/10.1038/s41598-019-43610-y PMID: 31076604 PMCID: 6510895
37. Meyerhardt J and Mayer R (2005) Systemic therapy for colorectal cancer N Engl J Med 352 476–487 https://doi.org/10.1056/NEJMra040958 PMID: 15689586
38. Wilson K, Fitzgerald C, and Barnett J, et al (2007) Adjuvant therapy with raltitrexed in patients with colorectal cancer intolerant of 5-fluorouracil: British Columbia Cancer Agency experience Cancer Invest 25 711 https://doi.org/10.1080/07357900701518388 PMID: 18058467
39. Bolzacchini E, Luchena G, and Giordano M (2019) Safety report of TAS-102 in a patient with reduced DPD activity Clin Colorectal Cancer 18 310–312 https://doi.org/10.1016/j.clcc.2019.07.008 PMID: 31421985
40. Henricks L, Siemerink E, and Rosing H, et al (2018) Capecitabine-based treatment of a patient with a novel DPYD genotype and complete dihydropyrimidine dehydrogenase deficiency Int J Cancer 142 424–430 https://doi.org/10.1002/ijc.31065
41. Ma W, Saif W, and El-Rayes B, et al (2016) Clinical trial experience with uridine triacetate for 5-fluorouracil toxicity (abstract) J Clin Oncol 34(suppl 4S) 655 https://doi.org/10.1200/jco.2016.34.4_suppl.655
42. von Borstel R, O’Neil J, and Saydoff J, et al (2010) Uridine triacetate for lethal 5-FU toxicity due to dihydropyrimidine dehydrogenase (DPD) deficiency (abstract) J Clin Oncol 28(15_suppl) e13505 https://doi.org/10.1200/jco.2010.28.15_suppl.e13505
43. Salem M, Nassef M, and Salam S, et al (2016) Effect of administration timing of post-chemotherapy granulocyte colony-stimulating factor on host immune cell recovery and CD8+ T-cell responses J Immunotoxicol 13 784–792 https://doi.org/10.1080/1547691X.2016.1194917 PMID: 27417188 PMCID: 5669798
44. Murphy C, Byrne S, and Ahmed G, et al (2018) Cost implications of reactive versus prospective testing for dihydropyrimidine dehydrogenase deficiency in patients with colorectal cancer: a single-institution experience Dose Response 16 1559325818803042 https://doi.org/10.1177/1559325818803042 PMID: 30288154 PMCID: 6168732
45. Cunha-Junior G, Bastos-Rodrigues L, and Azevedo P, et al (2019) Prevalence of the DPYD variant (Y186C) in Brazilian individuals of African ancestry Cancer Chemother Pharmacol 84 1359–1363 https://doi.org/10.1007/s00280-019-03974-4 PMID: 31641844
46. Conroy T, Hammel P, and Hebbar M, et al (2018) FOLFIRINOX or gemcitabine as adjuvant therapy for pancreatic cancer N Engl J Med 379 2395–2406 https://doi.org/10.1056/NEJMoa1809775 PMID: 30575490
47. Conroy T, Desseigne F, and Ychou M, et al (2011) FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer N Engl J Med 364 1817–1825 https://doi.org/10.1056/NEJMoa1011923 PMID: 21561347
48. Kadakia A, Rajan S, and Abughosh S, et al (2015) CMF-regimen preferred as first-course chemotherapy for older and sicker women with breast cancer: findings from a SEER-Medicare-based population study Am J Clin Oncol 38 165–173 https://doi.org/10.1097/COC.0b013e31828f5b01
49. Ministério da Saúde Incidência de Câncer no Brasil. Estimativa 2020 [https://www.inca.gov.br/sites/ufu.sti.inca.local/files//media/document//estimativa-2020-incidencia-de-cancer-no-brasil.pdf] Date accessed: 01/05/21
50. Ministério da Saúde Incidência de Câncer no Brasil. Estimativa 2020 – ERRATA [https://www.inca.gov.br/sites/ufu.sti.inca.local/files//media/document//estimativa-2020-errata.pdf] Date accessed: 01/05/21
51. World Health Organization International Agency for Research on Cancer [http://gco.iarc.fr] Date accessed: 01/08/21
52. Surveillance, epidemiology, and end results (SEER) – cancer stat facts: colorectal cancer [https://seer.cancer.gov/statfacts/html/colorect.html] Date accessed: 01/08/21




