Guidelines for the management of neuroendocrine tumours by the Brazilian gastrointestinal tumour group
Rachel P Riechelmann1,2,3, Rui F Weschenfelder4, Frederico P Costa3, Aline Chaves Andrade5, Alessandro Bersch Osvaldt4,6,7, Ana Rosa P Quidute8,9, Allan dos Santos3, Ana Amélia O Hoff1,3, Brenda Gumz3, Carlos Buchpiguel1,2, Bruno S Vilhena Pereira10, Delmar Muniz Lourenço Junior1,11, Duilio Reis da Rocha Filho9, Eduardo Antunes Fonseca3,12, Eduardo Linhares Riello Mello10, Fabio Ferrari Makdissi1,13, Fabio Luiz Waechter14, Francisco Cesar Carnevale2,3, George B Coura-Filho1, Gustavo Andrade de Paulo1,15, Gustavo Colagiovanni Girotto16,17, João Evangelista Bezerra Neto1,3, João Glasberg1, Jose Claudio Casali-da-Rocha18, Juliana Florinda M Rego19, Luciana Rodrigues de Meirelles3, Ludhmila Hajjar1,20, Marcos Menezes1,3, Marcello D Bronstein11, Marcelo Tatit Sapienza1,2, Maria Candida Barisson Villares Fragoso1,11, Maria Adelaide Albergaria Pereira11, Milton Barros21, Nora Manoukian Forones22, Paulo Cezar Galvão do Amaral23, Raphael Salles Scortegagna de Medeiros22, Raphael L C Araujo24, Regis Otaviano França Bezerra1, Renata D’Alpino Peixoto27,28, Samuel Aguiar Jr21, Ulysses Ribeiro Jr1,13, Tulio Pfiffer1,3, Paulo M Hoff1,2,3 and Anelisa K Coutinho26
1Instituto do Câncer do Estado de São Paulo, Universidade de São Paulo, São Paulo 01246-000, Brasil
2Departamento de Radiologia e Oncologia da Faculdade de Medicina da Universidade de São Paulo, São Paulo 01246-903, Brasil
3Hospital Sírio-Libanês, São Paulo 01308-050, Brasil
4Hospital Moinhos de Vento de Porto Alegre, Porto Alegre 90035-000, Brasil
5Oncocentro, Belo Horizonte 30360-680, Brasil
6Departamento de Cirurgia, Universidade Federal do Rio Grande do Sul, Porto Alegre 90040-060, Brasil
7Hospital de Clinicas de Porto Alegre, Porto Alegre 90035-903, Brasil
8Departamento de Fisiologia e Farmacologia da Faculdade de Medicina da Universidade Federal do Ceará, Fortaleza 60020-180, Brasil
9Hospital Universitário Walter Cantidio, Ceará 60430-370, Brasil
10Instituto Nacional do Câncer, Rio de Janeiro 20230-240, Brasil
11Disciplina de Endocrinologia e Metabologia, Faculdade de Medicina da Universidade de São Paulo, São Paulo 01246-903, Brasil
12 Department of Surgery, AC Camargo Cancer Centre, São Paulo 01509-010, Brasil
13Departamento de Gastroenterologia da Faculdade de Medicina da Universidade de São Paulo, São Paulo, Brasil
14Universidade Federal de Ciências da Saúde de Porto Alegre, Porto Alegre 90050-170, Brasil
15Hospital Albert Einstein, São Paulo 05652-900, Brasil
16Hospital de Base da Faculdade de Medicina de São José do Rio Preto, São Paulo 15090-000, Brasil
17Santa Casa de São José do Rio Preto, São José do Rio Preto 15025-500, Brasil
18Hospital Erasto Gaertner, Pontifícia Universidade Católica do Paraná, Curitiba 81520-060, Brasil
19Universidade Federal do Rio Grande do Norte, Natal 59300-000, Brasil
20Instituto do Coração, Universidade de São Paulo, São Paulo 05403-900, Brasil
21 Medical Oncology, AC Camargo Cancer Centre, São Paulo 01509-010, Brasil
22Disciplina de Gastroenterologia, Universidade Federal de São Paulo, São Paulo 04021-001, Brasil
23Hospital São Rafael, Salvador 41253-190, Brasil
24Departamento de Cirurgia do Aparelho Digestivo Alto e Hepato-Bilio-Pancreática, Hospital de Câncer de Barretos, São Paulo 14784-400, Brasil
25Departamento de Patologia da Faculdade de Medicina da Universidade de São Paulo, São Paulo 01246-903, Brasil
26Clinica AMO, Salvador 1950-640, Brasil
27Hospital São José, São Paulo 01323-001, Brasil
28Universidade Nove de Julho, São Paulo 02111-030, Brasil
Correspondence to: Rachel Riechelmann. E-mail:
Abstract
Neuroendocrine tumours are a heterogeneous group of diseases with a significant variety of diagnostic tests and treatment modalities. Guidelines were developed by North American and European groups to recommend their best management. However, local particularities and relativisms found worldwide led us to create Brazilian guidelines. Our consensus considered the best feasible strategies in an environment involving more limited resources. We believe that our recommendations may be extended to other countries with similar economic standards.
Keywords: neuroendcrine tumours, cancer, chemotherapy, targeted therapy, radionuclide peptide therapy, guideline
Copyright: © the authors; licensee ecancermedicalscience. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Published: 26/01/2017; Received: 09/11/2016
Introduction and methodology
The World Health Organization (WHO) categorises neuroendocrine tumours (NETs) according to the tumour prognosis, considering the histopathology and proliferative index of tumours [1]. However, NETs are heterogeneous and may present with different clinical forms and behaviours. In this context, international guidelines have proposed treatment algorithms to facilitate better clinical management. Regional particularities should also be considered, which inspired the development of a Brazilian consensus. This paper presents the findings and recommendations of a panel of Brazilian specialists in various medical fields on the diagnosis, staging and treatment of NETs. All topics for discussion were previously selected and distributed among the members. The presentations and polls of experts on the panel took place during face-to-face meetings. The medical literature related to the selected subjects was assessed using MEDLINE (National Library of Medicine). Scientific evidence and recommendations were classified according to the CDC classification system (Tables 1 and 2). In the absence of sufficient evidence to obtain a clear conclusion, the final recommendations were made by consensus and voting, based on the opinion of the panel experts.
Table 1: Levels of Evidence—CDC Grading System (Adapted from [127]).

Table 2: Strength of Recommendations—Classification System CDC (Adapted from [127]).

Diagnosis and staging
Pathology
NETs are tumours featuring neuroendocrine differentiation and may develop in various organs. The morphological classification proposed by WHO suggested three distinct prognostic groups: (1) grade 1 (G1) [Ki67 ≤ 2% and <2 mitoses/10 high-power fields (HPF)], (2) grade 2 (G2) (Ki67 3%–20% and 2–20 mitoses/10 HPF) and (3) grade 3 (G3) (Ki67 > 20% and >20 mitoses/10 HPF) [1]. G1 and G2 tumours are characterised as highly differentiated patterns with nested groups or pseudo-glandular arrangements. The poorly differentiated G3, which corresponds to the neuroendocrine carcinomas (NECs), may have a pattern of small or large cells.
Recommendations
• The pathology report should be used as the gold diagnostic standard.
• Immunohistochemistry (IHC) should be conducted for all suspected cases of NETs with a minimal panel of markers (low-weight cytokeratin, synaptophysin and chromogranin A) to confirm the neuroendocrine nature of the tumour [IVA], and reduce the chance of diagnostic errors.
• The proliferative index should be evaluated by counting mitotic figures using optical microscopy and the percentage of positive cells identified by IHC to determine the Ki67. The degree of determination requires a count of mitotically active cells in several areas, with at least 50 HPF (1 HPF = 2 mm2). The average of mitosis should be based on a count using 10 HPF. We recommend that the determined Ki67 should be expressed as a percentage of 500–2000 neoplastic cells counted in the areas of greater nuclear staining, known as hot spots. In a situation where a pathological analysis is performed using aspirated biopsy or a fine needle in which the cell count is below 500 cells, histologic grading is not recommended. Thus, the pathology report should be limited to confirming the neuroendocrine nature of the tumour.
• The proliferative index of Ki67 and/or mitotic cells can be done manually or with the aid of electronic programs for image analysis. The ratio of the percentage of positive cells over the total number of cells should be expressed in absolute numbers in the ICH report [2] [IVA].
• In the event of inconsistencies between mitotic count and Ki67, the higher histological grade should be used [1, 3] [IVA].
• Histologic degree of differentiation should be used to distinguish NETs as well differentiated (WD) or poorly differentiated, according to the architectural features (standard acinar, trabecular, solid, nests or diffuse) and cytomorphological (absence of pleomorphisms, small cells or large cells). We recommend that reporting the histologic degree of differentiation regardless of the proliferative index, i.e. NETs with a high proliferative index (>20%) should be described according to their cell differentiation and not solely as carcinomas [1, 3] [IVA].
• Poorly differentiated tumours should be classified in small or large cells [VB].
• Although there are many different staging systems for NETs, for example, the modified ENETS staging system for pancreatic NETs [4], for resected tumours, we recommend performing the pathological staging according to the guidelines in the American Joint Committee on Cancer, Seventh Edition [VA].
• Routine testing for other markers is not necessary [VE], except in cases of primary site determination where pancreatic markers (insulin, glucagon, etc) can guide the diagnosis of occult primary NETs [VB].
• Histological parameters should be described in all cases of primary tumour resection (i.e. surgical specimens, mucosectomias or excisions) [1–3, 5] [IVA].
Pathological reports should include
• Anatomical site;
• Diagnosis;Three-dimensional measurements of the lesion; Descriptions of unusual histology (oncocytic, clear cells, glands, etc);
• IHC results for neuroendocrine markers;
• Descriptions of multicentric disease;
• Descriptions of methods and systems used for grading;
• Mitotic index at 10 HPA or 2 mm, and counts at 50 HPA for hot spots;
• Descriptions of Ki67 indices in percentage values. A visual counting method is appropriate, but counts using printed images and/or by using software are preferable. Ki67 index, counting multiple hot spots for biopsies where the diagnosis of NECs cannot be excluded;
• Descriptions of vascular and perineural invasion;
• Descriptions of metastastatic lymph nodes, given as a ratio (number of affected nodes / number of examined nodes);
• TNM staging (refer to guidelines);
• Surgical margins, given as the distance between the tumour and the margin, if the distance is <0.5 cm;
• Proliferative changes or other abnormalities;
• Distance of invasion
◦ Stomach: Depth of invasion in the gastric wall;
◦ Small intestine: Depth of the intestinal wall invasion;
◦ Colon: Depth of invasion in the wall of the large intestine;
◦ Appendix: Depth of invasion in the wall of the appendix; description of presence of invasion extended to mesoappendix;
◦ Pancreas: Presence of extra-pancreatic extension or bile duct invasion, duodenum or ampoule;
◦ All sites: Involvement of serous or peritoneal surface, invasion of structures and/or adjacent organs.
• Presence of other non-neuroendocrine components;
• Presence of non-ischemic tumour necrosis.
Resection or biopsy of metastatic tumours
• Location and number of metastases;
• Extension of the involvement of the resected tissue, as a percentage;
• Dimensions of the largest metastasis;
• Presence of unusual histology (oncocytic, clear cell, glands, etc);
• IHC of neuroendocrine markers;
• Identification of the primary site by IHC;
• Grading;
• Mitotic index at 10 HPF or 2 mm; 50 HPF count;
• Ki67 index;
• Presence of other non-neuroendocrine components;
• Presence of non-ischemic tumour necrosis;
• For resected cases, describe the surgical margin as the distance between the tumour and the margin, if the distance is <0.5 cm.
Utility of hormones, peptides, chromogranin A, 5-HIAA, gastrin and insulin in the diagnosis of NETs
Chromogranin A
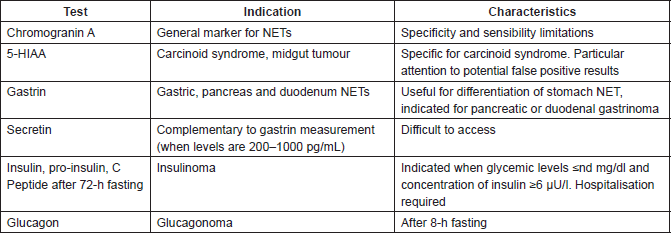
Chromogranin A is acceptable, despite its limitations, as a general marker of NETs. The sensitivity and specificity limitations of chromogranin A may depend on the test used and clinical situations, such as secondary hypergastrinemia, gastrinoma, atrophic gastritis, Helicobacter pylori infection, use of proton pump inhibitors and liver or kidney dysfunction. Elevated levels of chromogranin A may also be caused by other neoplasias, diminishing its specificity [6] (Table 3).
Recommendations
• Chromogranin A can be used as a prognostic test [IIIC], and can be used for monitoring purposes [7] [IVC]. It should not be used alone to guide management.
• Repeated chromogranin A measurements can be made to assess tumour response [8], although the results should not be used to determine management [IIIC].
Table 3: Test Indication.
5-hydroxindoleacetic acid
5-hydroxindoleacetic acid (5-HIAA) is a metabolite of serotonin associated with carcinoid syndrome, being useful for initial and follow-up evaluations. However, high levels of 5-HIAA are also observed in patients with celiac disease, intestinal stasis, cystic fibrosis and chronic use of drugs that enhance serum serotonin (serotonin reuptake inhibitors and tramadol), while low levels are observed in patients with impaired renal function (Table 3).
Recommendations
• Patients should receive a low-serotonin diet for 48 h prior to and during collection of 5-HIAA in the urine (24 h) to avoid overestimation, as well as the withdrawal of serotoninergic drugs (e.g. antidepressants), if possible [VA].
• The 5-HIAA in the urine (24 h) should be measured in patients with carcinoid syndrome [IIA] and in all patients with metastatic midGut NETs involving the liver, even in the absence of specific symptoms [IVB].
• Dosage is optional for prognostic purposes [9] [IIIC].
• For patients receiving somatostatin analogues (SAs), 5-HIAA should be measured immediately before the next dose, in order to avoid artificially low values. [VA]
Insulin
All recommendations are IVA
Measuring serum insulin, proinsulin and C-peptide following prolonged fasting (72 h) during hypoglycemia is considered the gold standard for diagnosis of insulinoma. However, the fasting test has limited application, because it requires hospitalisation to monitor blood glucose. In practice, documentation of clinical and laboratory hypoglycemia in the presence of Whipple’s triad accompanied by hypersinsulinemia is usually sufficient and recommended for diagnosing insulinoma.
The absolute values of glucose and insulin are the most relevant data, and any measurable insulin ≥ 3 mcU/ml ≥ 20, 8 pmol/l when blood glucose levels fall to values ≤ 55mg/dl or 3 mmol/l should be considerable abnormal [10, 11]. The final proinsulin dose in the fasting test shows high sensitivity and specificity for the diagnosis of insulinoma when concentrations are higher than 22 pmol/l. Patients with insulinoma show significantly lower, non-overlapping proinsulin levels compared with individuals without it [12, 13].
Gastrin
All recommendations are IVA
Gastrin levels are useful for differentiating various types of stomach NETs [14]. Type I: Good prognosis associated with atrophic gastritis, usually multiple, presence of hypergastrinemia, and vitamin B12 deficiency; Type II: uncertain malignant potential, usually multiple, associated with hypergastrinemia in patients with gastrinomas and/or multiple endocrine neoplasias (MEN-1); Type III: Aggressive, often isolated and gastrin normal levels. It is also important to determine gastrin levels in cases of suspected gastrinoma (Table 3). Provocative tests or stimulation with calcium or secretin may be indicated when the gastrin concentrations are between 200 and 1000 pg/ml [15]. Other tests for specific hormones secreted by NETs should follow clinical indications.
General Recommendations for Functioning NET
• We do not recommend hormonal screenings for all patients in the absence of clinical indications, except for urinary 5-HIAA in asymptomatic patients with liver metastatic midGut NETs [VA]. In these cases, the identification of subclinical elevations of this marker may be useful for monitoring carcinoid heart disease (CHD).
• In rare cases of atypical carcinoid syndrome and 5-HIAA concentrations are in the upper normal limits, platelet serotonin measurement may be useful [IVC].
Conventional imaging tests
The most commonly used conventional imaging tests for NETs are ultrasound (US), computed tomography (CT) and magnetic resonance imaging (MRI). US tests have extensive variability in the detection of pancreatic NETs (17%–79%), making this method unsuitable for identifying NETs [16]. However, endoscopic US (EUS) has shown an approximately 90% of detection rate for NETs in the head or pancreatic neck and duodenum [17]. CT studies with biphasic protocol showed 94% of sensitivity, and can detect pancreatic tumours at a rate similar to EUS. The combined sensitivity of the two methods has been described as 100% [18].
CT with multislice detector (CTMD) has an average detection capability of 73% (39%–94%) for pancreatic NETs [16]. False negative results can occur with tumours <2 cm and insulinomas. Thus, sequential evaluation with CTMD and EUS is recommended for pancreatic NETs, except in cases of suspected insulinomas [19]. CTMD with three-phase protocol is a useful tool for diagnosis and staging [20]. The EUS, 68Ga positron emission tomography (PET/CT68Ga) and CTMD have comparable accuracies in the diagnosis of duodenopancreatic NETs [21]. MRI has been found to be comparable with EUS in detecting duodenopancreatic NETs, and is recommended as the best method to use, employing the T2 and post-contrast T1 sequences in the arterial phase [22].
Recommendations
• US for primary assessment of NETs should not be used [16] [IVD].
• MRI, CTMD and EUS have a similar sensitivity to detect pancreatic NET [17, 18]. However, CTMD is preferred for surgical planning for better vascular assessment [20] [IVB].
• Three-phase CT is the preferred method for staging liver metastases from NETs [VA]. MRI can be used to evaluate liver metastases, if available [23] [IVB].
• CT enterography is optional and experimental [24] [IVC].
Endoscopic ultrasound for diagnosing pancreatic and duodenal lesions
The sensitivity of EUS for diagnosing duodenopancreatic NETs is 87%, and its specificity is 98% [25]. For insulinomas, the sensitivity is 87.5% and the specificity is 97.4%. [25]. For gastrinomas, the sensitivity is 84.5% and the specificity is 95% [25]. In addition to diagnostic use, EUS can also be used with fine needle aspiration for histological studies [26].
Recommendation
• EUS can be used for the diagnosis of pancreatic or duodenal NETs [25] [IIA].
PET/CT18F/FDG PET/CT68Ga-somatostatin analogue
The 18F-Fluorodesoxiglucose (18F/FDG) is a radiopharmaceutical analogue of glucose and PET/CT68Ga is an analogue (agonist) of somatostatin receptor type 2. While the performance of scintigraphy octreoscan is similar to multidetector CT [27], the accuracy of PET/CT68Ga is superior when compared with conventional imaging tests for WD NETs [28, 29]. The PET/CT18F/FDG has limited accuracy in staging WD NETs [30].
Recommendations for the use of PET/CT18F/FDG PET/CT68Ga-somatostatin analogue
• PET/CT18F/FDG can be used to stage for metastases of initially resectable G3 [31] [IVC], but should not be used to determine treatment.
• PET/CT68Ga-SA, if available, is indicated and preferable to octreoscan for staging G1 and G2 NETs [28, 29] [IIA].
• PET/CT68Ga-SA or octreoscan can be used to determine the somatostatin receptor expression. This is necessary for the indication of radiopharmaceutical therapy (see recommendations for radionuclide peptide therapy) [IIA].
• PET/CT68Ga-SA is recommended in cases of unknown primary lesions or suspicious lesions [32] [IIIA].
• PET/CT68Ga-SA can be used to evaluate suspected recurrences after surgery [33] [IIIA].
Whole-body magnetic resonance
Whole-body magnetic resonance (WBMR) employs three sequences: (1) STIR, (2) T1 (3) and diffusion. Its advantages include lower scanning costs and absence of ionising radiation or intravenous contrast administration. PET/CT68Ga apparently has superior sensitivity compared with WBMR for detecting WD NETs, specifically in bone and primary lesions [29].
Recommendations
• WBMR can be used to stage WD NETs when PET/CT68Ga is not available [29] [IIIC].
• WBMR, if available, is a method with high accuracy for screening patients with NETs without ionising radiation [34] [IIIB].
Extended examinations of unknown primary tumours
Patients with unknown primary tumours are characterised as such when conventional tests such as CT, MRI, functioning peptide dosage, endoscopy and colonoscopy cannot determine NET origin. The importance of identifying an occult primary NET relies on the potential to impact patient survival [35–37], for example, by indicating the need for a liver transplant.
Among the methods of examination, surgery itself is an option where the primary tumour can be identified intraoperatively [38]. Enteroscopy has low accuracy in detecting NETs of the small intestine (sensitivity of 33%) [39]. As for functional tests, PET/CT68Ga is considered an excellent imaging method for detecting occult primary tumours [40–43]. An observational study showed that PET/CT68Ga identified 59% of occult NETs [41]. If PET/CT68Ga is not available, the SRS (octreoscan or 99mTc-octreotide SPECT/CT) can be used [44], albeit with less accuracy.
Enterotomography can be considered in selected cases [45]. The use of endoscopic capsule may also be considered, although its sensitivity varies widely in the literature (45%–80%) for detecting NETs [46, 47].
Recommendations
• EUS can be used in cases of suspected primary tumours in the stomach, duodenum and pancreas.
• PET/CT18F/FDG can be used in cases of suspected high-grade primary tumours [VD].
• Surgery can detect primary tumours intraoperatively independently of preoperative location [VC].
• PET/CT68Ga is one of the most accurate methods for detecting WD occult NETs [32, 40–43] [IIIA].
• If PET/CT68Ga is not available, we suggest using the SRS (octreoscan or 99mTc-octreotide SPECT/CT) [IVB].
• Enterotomography can be considered, particularly in cases of suspected Crohn’s disease [45] [VC].
• Endoscopic capsule [47] [IVC] and enteroscopy [VC] can be considered for occult small bowel NET examinations. Be aware that endoscopic capsule may cause bowel obstruction up to 2% of cases [48].
• IHC in the investigation of occult NETs can be useful, especially in occult pancreatic NETs (see pathology section) [VC].
Patient follow-up
There are very few studies to guide the follow-up of patients with NETs. Most recommendations have been based on expert opinions [49].
Recommendations
• NET G3: Clinical assessment without radiological tests should be performed every three months for two years and every six months from the third to fifth year [VB]. Conventional imaging (CT and MRI) of the abdomen, pelvis and chest can be used in cases of suspected recurrence.
• Gastrointestinal and pancreatic NET G1 or G2 treated with R0/R1 resection: Clinical and imaging tests (CT and/or MRI) should be performed every 4–6 months for two years. After this period, perform annual examinations every 3–5 years or earlier if there is suspicion of relapse [VB].
• Tumours <1 cm require monitoring only in the presence of negative factors of prognosis, such as poorly differentiated histology or high mitotic index [IIIB].
• Functional tests such as octreoscan and PET/CT68Ga are not recommended for follow-up in patients with no evidence of disease as determined by conventional imaging [VE].
• Chromogranin A measurement alone is not recommended [IVE].
Follow-up for patients with evidence of metastatic disease receiving treatment or watchful wait
Because there is no level 1 evidence for the follow-up of patients with NETs, there is room for personalisation of frequency of follow-up visits and imaging tests.
• Conventional imaging tests (CT and/or MRI) should be performed every 3–6 months [VA].
• In cases of rapid disease progression or dubious examination results, a new liver biopsy may be considered to reassess proliferation [VC].
Follow-up for patients with specific NETs
Gastric
• Type 1: Endoscopic assessment every 12 months in recurrent patients and every 24 months in non-recurrent patients [VB]. There was no consensus on the follow-up period. The follow-up should be based on clinical judgments in individual cases.
• Type 2: Consider the same recommendations for multiple endocrine neoplasia (see MEN section).
• Type 3: CT thorax, pelvis and abdomen should be performed every 3–4 months for the first two years and every 6–12 months from the third to fifth year [VB].
Colon and rectum
• Colorectal NETs <1 cm G3 and G1–G3 from 1 to 2 cm: Annual monitoring according to the protocol for adenomatous polyps [50].
• For cases of NETs >2 cm, we recommend annual colonoscopies for five years.
• Octreoscan or PET/CT68Ga should not be included in the routine follow-up.
Surgical treatment
Resection of the primary tumour: resectable disease
Recommendations
• We recommend complete resection with curative intent for all NETs.
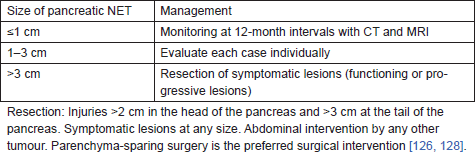
• ForWD pancreatic NETs <2 cm, particularly G1 tumours, we recommend surveillance, with CT scan performed every six months [IVB]. For growing tumours and/or those with higher ki67 index, surgery is recommended [51–53] [IVB].
• Midgut NETs should be treated with resection of mesenteric lymph nodes in order to prevent mesenteric fibrosis [VB].
Resection of the primary tumour in metastatic disease
The practice of resection of the primary NET in the metastatic setting is based on small retrospective series. In general, patients with good performance status and symptomatic primaries have indication for resection. Patients with liver transplant indications should also have the primary tumour resected. In asymptomatic metastatic patients, surgery for primary G1 or G2 tumours of the small intestine should be considered for the risk of obstruction, mesenteric fibrosis and intestinal ischemia [54].
Recommendations
• Resection of G1 and G2 midgut primary symptomatic NETs, with or without involvement of mesenteric and symptomatic primary colon or rectum NETs [55] [IVA].
• Resection of the pancreatic or gastric NET primary in the metastatic disease setting is not recommended [56] [IVE].
• Resection of asymptomatic primary tumours of the midgut can be considered in select cases, where the tumour presents a significant risk for complications such as obstruction and mesenteric fibrosis [54] [IVC].
Surgical treatment of liver metastasis: recommendations for resectable disease
Patients with G1 and G2 NETs presenting with metastases limited to the liver may be considered for resection of entire tumour. In order to obtain R0 resection, combined radiofrequency ablation should be considered for local control [57]. In general, resectable liver lesions are those that can be extracted with free margins and are maintaining at least two contiguous liver segments and their vascular pedicles, and biliary and venous drainage. In these cases, parenchymal preservation techniques such as portal embolisation [58] and staged hepatectomy [58] should be discussed with an experienced surgical team. While retrospective series have reported encouraging survival rates for NET patients undergoing hepatectomy [59, 60], overestimation for selection bias should be considered.
Recommendations
• The decision for liver resection of metastatic WD NETs should be discussed by multidisciplinary teams [VA]. R0 resection should be aimed whenever possible [57] [IVB].
• Ablative techniques such as radiofrequency can be combined with surgery in order to obtain negative margins. Intraoperative US [IVA] should be performed for staging liver lesions [57].
• Liver resection should be performed by experienced teams [VA].
• A simultaneous approach to primary and liver tumours can be considered [61] [IVB].
Surgical treatment of liver metastasis: recommendations for unresectable liver predominant disease
Debulking surgery
Debulking is indicated for functioning NETs to alleviate symptoms not adequately controlled with systemic therapy and/or locoregional therapies of the liver [62–64]. The magnitude of debulking surgery is arbitrary but considered by most as at least debulking >90% of liver lesions. The debulking of extrahepatic metastatic disease is not recommended. If the patient has CHD, heart surgery must be performed before surgical debulking for the high risk of intraoperative bleeding.
Recommendation for surgical debulking
• Recommended for patients with good performance status, functioning WD NETs, with predominant liver disease and poorly controlled clinical syndrome despite systemic and/or locoregional therapies [62, 64] [IVB].
Locoregional treatment
Embolisation and chemoembolisation
Patients with WD NETs limited to the liver or predominantly unresectable hepatic disease can benefit from locoregional therapies, including liver embolisation, combined or not with chemotherapy and percutaneous radioablation (with radioisotopes, cryoablation or radiofrequency). Contraindications for hepatic embolisation are prior pancreatic and/or biliary surgery, portal vein thrombosis and those with moderate-to-severe liver dysfunction.
Retrospective studies have evaluated the benefits of embolisation with reported symptomatic responses in 80% of cases and objective responses in approximately 30% [65]. Although there are no controlled studies comparing embolisation with chemoembolisation, we prefer chemoembolisation for pancreatic NETs, given their higher sensitivity to chemotherapy in comparison with midgut tumours. The most frequently used drugs in chemoembolisation are doxorubicin, mitomycin and cisplatin. Common symptoms of post-embolisation and post-chemoembolisation syndromes include nausea, vomiting, increased liver enzymes, abdominal pain and fever, and myelosuppression if combined with chemotherapy. Fever may persist for up to 30 days depending on the extension of tumour necrosis, also characterised as an inflammatory response to trauma. It is important to consider that patients with moderate hepatic impairment, previous reconstruction of the biliary tract or portal vein obstruction are contraindicated for liver embolisation, with or without chemotherapy, for the high risk of liver impairment and infections.
Recommendations
• Liver embolisation and chemoembolisation are safe and are indicated in symptomatic inoperable metastatic liver disease [65] [IIB] or asymptomatic, but progressive, disease [66] [IIIC].
• We prefer pure embolisation for midgut G1 NETs, limiting the addition of chemotherapy to cases of G2 pancreatic or G2 gastrointestinal NETs [VB].
• Both embolisation and chemoembolisation may be repeated, if no significant liver dysfunction is detected [VB].
Radiofrequency ablation
Radiofrequency ablation is associated with symptomatic and/or radiological response in 70%–80% of NET patients, with an overall five-year survival rate of 50% [67]. Radioblation may be performed by a percutaneous approach and can be considered for patients with inoperable diseases limited to the liver, with lesions <3–4 cm. Radioablation may also be considered intra-operatively, with the goal of achieving R0 surgery.
Recommendations
• Percutaneous radiofrequency ablation can be considered for palliation of WD inoperable and progressive NET patients with a low volume of liver metastasis [67] [IVB]. The best results can be achieved in patients with up to 5 nodules (<3 cm).
• Ablation with open radiofrequency can be used as a complement to surgery, in an attempt to achieve free margins and/or manage lesions with difficult resectability [57] [IVC].
Radioembolisation
Radioembolisation is a new transcatheter therapy that uses 90Yttrium as the radiation-emitting radioisotope. 90Yttrium releases beta radiation (β) with an average tissue penetration of 2.5 mm and a maximum penetration of 11 mm. Radioembolisation offers doses at 100–300 Gy, which would not be possible through external radiotherapy due to liver toxicity [68]. Post-embolisation syndrome affects 40%–50% of patients [65] and, in most cases, has low morbidity. Serious side effects, such as hepatitis, gastritis, peptic ulcers, pancreatitis, cholecystitis and pneumonitis, are rare, occurring in 2%–8% of cases. Hepatic disease induced by radiation is even more rare (<1%). In G1 and G2 NETs, retrospective studies show varying results for disease control [69, 70].
Recommendation
• Radioembolisation with 90Yttrium can be used as a rescue treatment for progressive liver disease, but should be used only in select cases of patients with G1 and G2 NETs, who have good hepatic reserve, and have been previously treated with locoregional and systemic therapies [69, 70] [IIIC].
Unresectable disease limited to liver: liver transplant
Liver transplantation may be a potentially curative option for selected patients with WD NET. Eligible cases for transplantation, however, should be very well selected [71–74].
• Liver transplantation may be considered in select cases for patients with G1 and G2 NETs, preferably with Ki67 < 10%, at least six months of stable, good performance status, minimum comorbidities, no extrahepatic disease and preferably when other treatment options have been exhausted [71, 73] [IVC].
• Assessments to rule out extrahepatic diseases should include total abdomen CT/MRI, chest CT, octreoscan or preferably, if available, PET/CT68Ga [VA].
• Eligible patients must have the primary tumour resected prior to transplantation.
Systemic treatment of gastroenteropancreatic NET
Somatostatin analogues as an anti tumour therapy versus watchful waiting
Two phase III placebo-controlled studies have demonstrated the antitumour effects of SAs (octreotide and lanreotide) in WD gastroenteropancreatic (GEP) NETs with Ki67 < 10% [75, 76], regardless of NET functionality.
Recommendations
• SAs are the first-line systemic treatment of choice for WD, inoperable and advanced progressive WD GEP NETs, with Ki67 < 10% [75, 76] [IB].
• We favour the confirmation of SSTR2 expression through octreoscan or PET/CT68Ga prior to SA-based treatment [IIB], although this is not mandatory [77].
• We consider octreotide and lanreotide to have similar safety and efficacy for treating WD GEP NETs [VB].
• Watchful waiting can be recommended in WD NETs, preferably those with G1, non-functioning, low-volume, and oligosymptomatic disease [VC]. WD G1 NETs are indolent and may stabilise for months to years without treatment as observed in the median of progression-free survival (PFS) in the placebo arm of the CLARINET study [75]. Furthermore, there is no evidence that SAs either have an impact on overall survival (OS) or on quality of life in patients with non-functioning tumours.
Interferon-α
Despite its unfavourable toxicity profile, interferon-α is still used and, in many low-resource countries, it is one of the few available treatments for NET. Several studies have shown that interferon-α provides tumour control, mostly through disease stabilisation and symptomatic response in carcinoid syndrome [53]. However, randomised studies have not shown that interferon-α alone or combined with SA was superior to monotherapy [53].
Recommendation
• Interferon-α can be used in cases of SA-resistant and progressive WD NETs [53, 78] [IC].
Chemotherapy for WD NETs
Chemotherapy, although not studied in phase III trials, is considered an effective treatment for G1 and G2 pancreatic NETs, with the response rates of 30%–40% with temozolomide and capecitabine [79], capecitabine and oxaliplatin [80], FOLFOX, dacarbazine or streptozotocin alone or associated with 5FU. The best regimen or the number of cycles has not been established, and this decision should consider limiting toxicities and maximum response. Chemotherapy has a limited role in patients with WD metastatic midgut NETs [79]. Predictive factors for chemotherapy responses have been studied. Methyl-guanilmetiltransferase (MGMT) deficiency as assessed by IHC or by methylation may have a predictive role for determining the alkylating response [81]. However, the best MGMT measurement method has not been determined. The role of Ki67 as a predictor of response to chemotherapy in WD NET is not established, but may be considered for chemotherapy indication.
Recommendations
• Patients with radiological and/or symptomatic progressive WD pancreatic NET [80] [IIB].
• Patients with WD gastrointestinal NET who have exhausted other treatment options [80] [IID].
◦ Suggested regimens: Capecitabine combined with temozolomide or oxaliplatin, FOLFOX and streptozotocin with or without 5FU.
• Increased expression of Ki67 can be used to recommend chemotherapy in WD GEP [VB].
• MGMT expression should not guide the use of temozolomide/dacarbazine [81] [IVD].
Targeted therapy in non-pancreatic gastrointestinal NET
Everolimus
Everolimus, an oral inhibitor of mTOR, was effective in WD pancreatic, gastrointestinal or pulmonary NETs. The RADIANT-2 study [82], a phase III, placebo-controlled, double-blind study of patients with WD functioning gastrointestinal and pulmonary NETs, found that 10 mg of everolimus continuously combined with 30 mg of intramuscular octreotide LAR prolonged PFS (median: 11.3 months to 16.4 months; hazard ratio (HR): 0.77; IC 95%: 0.59–1; p = 0.026). This value was not statistically significant by radiology central review, but suggested benefit by local evaluation. The benefit in PFS in gastrointestinal and pulmonary NET patients treated by everolimus was formally demonstrated by the RADIANT-4, a phase III trial of similar design, which included only patients with non-functioning tumours [83].
Recommendations
• Everolimus monotherapy is recommended for non-functioning and progressive gastrointestinal or pulmonary WD NETs [IA]. Everolimus may be used as a first-line or second-line treatment after SA [83] [IIB].
• Everolimus in combination with SA is recommended for patients with functioning tumours [IIA]. Utilisation is preferable after progression on SA monotherapy [82].
• Everolimus can be used as a monotherapy in functioning tumours to control hormonal syndromes [78, 84] [IVC].
Sunitinib
Sunitinib is an oral inhibitor of tyrosine kinase receptors, including PDGFR, VEGFR and c-KIT. A phase II study showed limited efficacy for sunitinib in treating gastrointestinal NETs, with the median PFS rate of 7 months in carcinoid tumours [85].
Recommendation
• Sunitinib is not recommended for patients with gastrointestinal non-pancreatic NETs [85] [IID].
Targeted therapy in WD pancreatic NET
The phase III placebo-controlled RADIANT-3 trial for patients with advanced, progressive pancreatic NET showed significantly prolonged PFS in favour of everolimus, with a median of 11 months versus 4.6 months (HR: 0.35; IC 95%: 0.27–0.45; p < 0.0001) [86]. Similarly, a placebo-controlled phase III study of sunitinib for the same population WD demonstrated the median PFS rate of 11.4 months versus 5.5 months (HR = 0.42; IC 95% = 0.26–0.66; p < 0.001) [87].
In pancreatic NETs, the choice between the two drugs should consider their toxicity profiles. Serious adverse events (G3 and grade 4) most commonly associated with sunitinib were neutropenia (12%), hypertension (10%), and hand and foot syndrome (6%), while everolimus commonly promoted stomatitis (7%), anemia (6%) and hyperglycemia (5%) [83, 87]. In a meta-analysis of individual data, severe pneumonitis related to everolimus occurred in approximately 2% of patients [88]
Recommendations
• Everolimus and sunitinib monotherapy are recommended for WD functional or non-functional, progressive advanced NETs [86, 87] [IA].
• Both drugs can be used in first-line therapy or following SA and/or chemotherapy [86, 87] [IIIA].
• Everolimus and sunitinib can also be combined with SA to treat functional tumours [86, 87] [IIA].
• In metastatic insulinomas, the use of everolimus is preferred drug [IVA].
Radioisotopes
Radioisotopes, also termed peptide radionuclide receptor therapy (PRRT), have been successfully used to treat patients with metastatic WD NETs, with somatostatin receptor positive expression confirmed by scintigraphy with 111Indium-labeled octreotide or PET/CT68Ga [89]. Contraindications for PRRT include pregnancy, severe psychiatric disorders, moderate or severe renal impairment and low bone marrow reserve.
In a large retrospective series of 500 patients with pancreatic or gastrointestinal NET treated with 177Lutetium, the overall response rate was 18% in the intention to treat population, with the median PFS rate of 33 months [90]. Patients were treated with maximum cumulative dose of 800 mCi. Treatment was well-tolerated with acute toxicity within 24 h of application that included nausea, vomiting and abdominal pain. Delayed toxicities may occur, particularly with PRRT with 90Itrium, with the most common events including hematologic (secondary myelodysplasia or leukemia), kidney failure and liver toxicity [89].
A recent study (NETTER-1) randomised 229 patients with G1 or G2 progressive midgut NET after SA to receive 60-mg octreotide LAR or 177Lutetium associated with 30-mg octreotide LAR [91]. Outcomes favoured PRRT, with the response of 19% versus 3%, median PFS not reached (estimated at 40 months) versus 8.4 months (HR: 0.2; p < 0.0001) and preliminary data suggests increased OS.
Recommendations
• 177Lutetium is recommended for progressive WD midgut NET [79] [IA].
• The best timing to administer 177Lutetium has not been established, although improved PFS has been demonstrated in the second-line setting. For limited data on delayed safety analysis of this treatment and perceptions of greater toxicity of chemotherapy and targeted-therapy after 177Lutetium, we prefer to indicate it after other systemic therapies have failed [VB].
• 177Lutetium is recommended for patients with an advanced WD pancreatic or hindgut NET whose disease progressed with SA, locoregional and/or systemic therapy [90] [IIIB].
• It is necessary to confirm SSTR2 expression by octreoscan or PET/CT68Ga prior to PRRT [79, 89, 90] [IA].
Management of bone metastases
Bone metastases occur in 18%–46% [92, 93] of WD NET and are associated with worse prognosis [94]. For management, we recommend specific treatments of NETs plus bisphosphonate or denosumab [95] at the physician’s discretion.
Recommendations
• There is no preferred imaging method for the diagnosis of bone metastases. Although PET/CT68Ga is the most accurate imaging method to stage bone metastases in WD NETs [96, 97], different imaging techniques can be used, such as bone scintigraphy and/or octreoscan, or whole body diffusion MRI [IVB].
• Bisphosphonates or RANK ligand inhibitors, such as denosumab, can be used in patients at risk for complications from bone metastasis [95] [IVC].
Management of hormonal syndromes and its complications
De novo and refractory carcinoid syndrome
Control of the carcinoid syndrome is important not only for the improvement of symptoms and consequently the patient’s quality of life, but also to prevent or delay the development of complications associated with carcinoid syndrome, such as retroperitoneal fibrosis and carcinoid cardiopathy [98]. SAs have been the first-line standard treatment of carcinoid syndrome for decades. However, symptomatic progression occurs, and various therapies have been tested in small phase II studies or retrospective series to treat refractory carcinoid syndrome [78]: (1) dose-escalation of SA, (2) interferon-α, (3) everolimus, (4) radioisotopes, (5) locoregional therapies and (6) liver debulking surgery. Recently, an oral inhibitor of the serotonin synthesis, telotristat etiprate, has shown better control of number of bowel movements per day in a phase III placebo-controlled trial [99]. The drug is not yet approved, and although current formal recommendations are difficult to make, it is likely that telotristat will be placed early in the treatment sequence of refractory carcinoid syndrome [78]. Chemotherapy has limited effects in midgut tumours, which are the most common type associated with carcinoid syndrome.
Recommendation for the treatment of carcinoid syndrome de novo
• We recommend the use of SA [IA]. We consider octreotide and lanreotide interchangeable for controlling carcinoid symptoms or other symptoms associated with hormonal syndromes in NET.
Recommendations for the management of carcinoid syndrome uncontrolled with SA label dosages.
• Physical examination should be performed to discard absorption problems (e.g. fibrosis in application site) [VA].
• Increased doses of SA can be used [78] [IIB]. There was no consensus on the recommended strength. There is little evidence to recommend the use of higher doses (>40 mg) of octreotide LAR. There is no evidence to support the use of increased doses of lanreotide (120 mg or more).
• Shorter intervals between injections for patients whose symptoms recur before next injection [VA]
• Everolimus [IVC] or associated with SA [78] [IIC].
• Interferon-α combined with SA [78] [IIIC].
• 177Lutetium or 90Itrium radioisotopes can be indicated for NETs with proven expression of SSTR2 [IVB] or I123-metaiodobenzylguanine (MIBG) in NET with proven expression of MIBG [78] [IVB].
• Surgical debulking of the liver [78] [IVB].
• Locoregional therapies such as embolisation of hepatic artery or radiofrequency ablation in liver predominant disease [78] [IIIB].
Glycemic control in insulinoma
Hypoglycemia caused by insulinoma can be fatal. Therefore, glucose control is extremely important, both in the preoperative period (when the vast majority of insulinoma are benign) and unresectable, or metastatic setting. Patients with insulinoma should be hospitalised for management of hypoglycemia.
Recommendations
• Hospitalisation for control of glycaemia with frequent meals at least every 3 h, continuous infusion of dextrose and glucose monitoring.
• Diazoxide for glycemic control [VB] can be used. However, this drug is not widely available.
• SA can be used cautiously for control of glycemia [VB]. This approach should be done carefully as regulatory hormones may be blocked leading to severe hypoglycemia. SA is recommended when SST2 expression is confirmed. Treatment should be initiated with short-acting octreotide for few days to better assess glycemic control. If effective, long-acting SA may be used.
• Everolimus has antitumour and hyperglycemic effects in metastatic insulinomas [84] [IVA].
• Verapamil and glucocorticoids are optional in selected refractory cases [VC].
• Treatment with targeted therapy or chemotherapy should be performed in combination with glycemic support [VA].
Management of carcinoid heart disease
CHD is among the most dramatic complications from carcinoid syndrome, as it is associated with poor prognosis [98]. The peptides released into the blood, particularly serotonin and other tachinines, mainly affect the right-hand side of the heart, damaging the tricuspid valve in 97% of cases [98]. Echocardiography with Doppler is the recommended diagnostic exam, allowing for the observation of mild regurgitation of the mitral valve, dilation of the right chambers and tricuspid valve insufficiency.
Recommendations
• We recommend screening with echocardiography in patients with elevated 24-h urine 5-HIAA, regardless of carcinoid symptoms [VA].
• The echocardiogram should be performed by a professional familiar with CHD [98] [IIIA], and this should include evaluation of patency of forame ovale by the bubble test. The measurement of NT pro-BNP, a predictor of right-hand-side heart overload is optional [98] [IIIC].
• In patients without CHD, an echocardiogram should be performed annually or when there at onset of symptoms of cardiac congestive failure [VA].
• Detailed assessment of the CHD severity can be performed by other diagnostic modalities (MRI, etc) [100] [IVC].
• Patients with CHD should be managed by a multidisciplinary team of oncologists, cardiologists, endocrinologists and surgeons [VA].
• Pharmacological and non-pharmacological treatment of heart failure, such as water and salt restriction, diuretics and digitalic drugs, can improve symptoms but do not alter clinical outcomes [VB].
• The definitive treatment of CHD is surgical valve replacement [IIA]. Surgery should be considered if the patient experiences symptoms of heart failure, right ventricular dilatation and decline of right ventricular function [98, 101]. Patients with poor performance status, metastatic disease and/or difficult to control carcinoid syndrome should not be considered candidates for valve replacement [IID].
• Valve repair, compared with valve replacement, should be avoided because of the risk of post-repair stenosis and significant valve damage associated with underlying carcinoid syndrome [IVD].
• Valve replacement, when indicated, should be performed in centres with experience in the treatment of NETs [VB].
• The use of biological valves (bioprosthesis) is preferred for the lower risk of bleeding compared with metallic valves [98, 101] [IVB].
• Avoid the use of opioids, neuromuscular inhibitors, adrenaline, noradrenaline, dopamine and isoproterenol during anesthetic induction in patients with CHD.
• We recommend the use of intraoperative IV octreotide beginning at least two hours before surgery, with continuous infusion for 48 hours after surgery [IIIB] with the goal of preventing carcinoid development [98].
Clinical management of glucagonomas, gastrinomas and vipomas
Glucagonoma
Glucagonoma is an aggressive pancreatic NET of glucagon-producing cells that often present with metastatic disease. Clinical manifestations include anemia, weight loss, diabetes and dermatological features of a typical necrolytic migratory erythema. They may also present stomatitis, glossitis, diarrhea, abdominal pain, psychiatric disorders and venous thromboembolism.
Recommendations
• Surgical treatment, if resectable, and the use of SA as a symptomatic treatment of clinical syndrome of glucagonoma.
• Consider parenteral nutrition, vitamin supplementation, assessments of the presence of depression and prophylactic anticoagulation in all patients with glucagonoma [VB].
Gastrinoma
Gastrinomas are characterised by ectopic secretion of gastrin from pancreatic or duodenal NET, and resultant gastric ulcers. Severe peptic ulcers with gastroesophageal reflux and diarrhea are characteristic of Zollinger–Ellison syndrome.
Recommendations
• We recommend the use of high doses of proton pump inhibitors, with or without H2 receptor blockers [102] [IVA].
• SA [VA] as an antitumour therapy can be used to control associated diarrhea.
Vipoma
Vipoma is a rare pancreatic NET that produces vasoactive intestinal peptide, an important peptide in the neuromodulation of intestinal function. Clinical manifestations include intense watery (choleric) diarrhea, with fluid and electrolytes depletion, risk of acidosis and hypovolemic shock, reductions gastric acid secretion, hyperglycaemia, hypercalcemia and flushing.
Recommendation
• We recommend SA as the first treatment [IVA]. It is important to manage blood volume, hypocalcemia and acidosis [103].
Neuroendocrine carcinomas or G3
NECs or G3 are rare and associated with poor prognosis, with a median OS of less than a year [104]. Retrospective studies suggest OS gains with adjuvant therapy [105]. For metastatic disease, platinum-based chemotherapy is considered standard [104], without apparent clinically relevant differences between cisplatin and carboplatin [106]. Recently, retrospective series have shown that G3 tumours may present WD histologies and that this subgroup has a lower proliferative index and better prognosis compared with poorly differentiated carcinomas [107, 108].
A retrospective multicentre European study showed higher Ki67 index is associated with better response to platinum-based chemotherapy [106]. Ki67, however, was not found to be a predictor for response to chemotherapy in other studies [108].
Localised G3
Recommendations
• Resectable G3 NEC should undergo oncological surgery [105] [IVA].
• Consider definitive treatment with platinum-based chemotherapy combined with radiotherapy for locally advanced or unresectable tumours [105] [IVA].
• Adjuvant chemotherapy with cisplatin (or carboplatin) associated with etoposide or irinotecan for 4–6 cycles can be considered in patients with good performance status after surgical resection [105] [IVC].
Treatment of metastatic disease
Recommendations
• Cisplatin (or carboplatin) associated with irinotecan or etoposide for first-line treatment [104, 106] [IIIA].
• Cisplatin (or carboplatin) may be offered again to responders who have received their last dose more than three months before [VB].
• Temozolomide-based or oxaliplatin-based regimens may be used in WD G3 tumours [VC].
• Temozolomide or dacarbazine can be used as second-line treatments after platinum regimens [109] [IIIB].
Common hereditary syndromes associated with GEP NETs: multiple endocrine neoplasia type 1 (MEN-1) and von Hippel–Lindau syndrome
GEP NETs may be associated with hereditary syndromes as MEN-1, von Hippel–Lindau (VHL) and neurofibromatosis-1, among others. In this consensus, we have focused on MEN-1 and VHL as they are the most commonly associated syndrome with NETs.
MEN-1: clinical and molecular diagnosis
The inheritance pattern of MEN-1 is autosomal dominant with almost complete prevalence by 50 years of age. Patients with MEN-1 have high susceptibility to the development of tumours in the pituitary gland, the parathyroid glands, and duodenal endocrine cells or pancreatic islets [110]. The clinical diagnosis of MEN-1 involves tumours located in at least two of the three main sites (pituitary, parathyroid and pancreas/duodenum). The diagnosis of familial MEN-1 is defined beyond a clinical diagnostic index case of MEN-1 when a first degree relative presents a tumour diagnosis in at least one of the three main sites [111–113].
Genetic diagnosis is made in individuals with a MEN1 germline mutation, whether or not they have any clinic manifestations related to MEN-1. However, it is essential that genetic counselling is carried out and that the DNA collection for mutation research on the MEN1 gene is offered to all patients with the clinical diagnosis of MEN-1. Mutations not identified by Sanger sequencing analysis should be sought by deletion analysis by multiplex ligation-dependent probe amplification (MLPA) [113]. In specialised centres, it is possible to proceed with the investigation of other genes (p15, p18, p21, p27 and AIP) in patients who are negative for the presence of mutation by the aforementioned techniques [113]. Unlike the fully proven benefit-risk genetic research value of the MEN1 gene by Sanger sequencing and MLPA, the clinical value of analysing these other MEN-1-related genes has not yet been established, because they are exceptionally rare.
Considering that MEN-1 is a rare disease, the level of evidence for the recommendations is based on expert opinion [VA].
• Genetic analysis of the MEN1gene should be offered to symptomatic and asymptomatic first-grade relatives of the index case with a known mutation.
• Mutation detection in symptomatic cases and MEN-1-related tumours are important to exclude phenocopies (relatives who develop sporadic MEN-1-related tumours and, therefore, are negative for mutation found in the family). Phenocopies should receive genetic counselling, being informed of the absence of risk of disease transmission to descendants. In addition, the definition of phenocopies can result in change of surgical and clinical management, which follows the standard recommendation for sporadic cancers.
• We recommend clinical screening with hormonal and radiological tests for all patients with MEN-1 and asymptomatic family members recognised as having MEN1 germline mutations. Early diagnosis allows for the best surgical treatment and potentially promotes increased survival and quality of life in patients with mutation.
• Clinical screening and laboratory tests of patients and asymptomatic carriers of MEN1 mutation should be performed annually.
• Annual hormonal examinations for pituitary tumours should be initiated at 5 years old with prolactin measurements, growth hormones and IGF1, along with active clinical examinations for symptoms associated with insulinoma.
• The beginning of primary hyperparathyroidism (HPT) investigations should be initiated at 8 years old with total calcium dosage, ionised calcium and parathyroid hormone measurements.
• Active investigation of clinical symptoms of gastrinoma should be made beginning at 10 years of age. For asymptomatic cases, gastrin dosage should be done annually at 20 years old of age.
• Radiological investigations, including MRI of the pituitary, MRI of the abdomen and chest CT should be made from 5, 10 and 15 years old, respectively, for the investigation of pituitary non-functioning adenoma, non-functioning pancreatic tumours and bronchial/thymic tumours. The frequency of radiological images is not clearly defined, but we recommend that they be provided every 1–3 years.
• Analysis of the MEN1 gene should be offered and extended to patients with atypical presentation of MEN-1, cases of suspected MEN-1 that do not meet clinical criteria (e.g. HPT association and thymic carcinoid tumour), young patients diagnosed with HPT for adenoma (<30 years) or parathyroid hyperplasia (<40 years) or multifocal pancreatic NETs.
• Patients with isolated gastrinoma may be investigated for association with primary HPT and family history of MEN-1, although the cost-effectiveness of genetic testing all cases, independently of age, is unknown.
• Patients with MEN-1 should be managed by an experienced multidisciplinary team.
MEN-1: management of localised disease
Gastrinomas associated with MEN-1 are generally small, multiple, located in the duodenum and indolent, especially when lesions are <2 cm [114, 115]. The management of gastrinoma is mainly focused on the control of symptoms related to hypergastrinemia. The use of high doses of proton pumps inhibitors (e.g. omeprazole, 80–160 mg/day) is usually effective in controlling symptoms.
Most insulinomas are benign but must be found and operated regardless of their size. The goal of surgery is not disease control, but to control the clinical manifestations related to hyperinsulinemia [116].
Recommendations
• We recommend surgical exploration in cases of gastrinomas >2 cm, preserving pancreatic function if possible. The goal of surgery is to control disease progression and metastasis.
• We recommend surgery for all cases of localised insulinoma, with transoperative control of hypoglycemia (US with dosages of insulin and glucose).
• We recommend monitoring non-functioning pancreatic NETs <2 cm using conventional screening images at intervals of 6 months to 1 year [117, 118]. For tumours >2 cm, we recommend surgical intervention [113] [IVB].
• For patients with MEN-1-associated metastatic pancreatic NET, we prefer everolimus as the first option [119] [VC].
MEN1: management of pituitary
Pituitary adenomas
The pituitary adenomas constitute about 15% of all intracranial tumours. The classification is morphological: microadenomas are <1 cm and macroadenomas are >1 cm. Both can be invasive, but pituitary tumours associated with MEN-1 tend to be more aggressive than sporadic forms. Adenomas can be secreting or not.
Recommendations
• The first choice of therapy for prolactinomas should include dopamine agonists, and cabergoline is the preferred medication [120, 121] [IA].
• Surgical treatment is recommended in cases of resistance or intolerance to dopamine agonists [121] [IVA].
• Temozolomide is indicated for rare cases of aggressive and resistant tumours [122, 125] [IIIB].
• Treatment with radiation therapy may be considered in tumours with local complications [121] [IVB].
• Repeated surgical resection can be performed in recurrent tumours [IVB].
Acromegaly
Acromegaly is a rare disease, but with high morbidity and mortality rates. Surgery is the primary treatment for most cases. SA, in particular, dopaminergic agonists and GH receptor antagonists comprise the treatment for cases that are not controlled surgically or cases with restrictions to surgery. Radiation therapy has been less often used.
Recommendations
• Surgery is the first treatment option [122].
• We recommend the use of SA to treat symptoms and signs [IB]. Both octreotide and lanreotide are clinically equivalent [122].
• Pegvisomant can be used in SA-refractory cases [123, 124] [IB].
Cushing’s disease
Recommendations
• Trans-sphenoidal surgery of the pituitary gland is the first choice of therapy [122] [IIA].
• We recommend drug therapy with ACTH modulators or blockers of adrenal synthesis of cortisol and/or radiation therapy for cases uncontrolled by surgery [122] [IVB].
• Bilateral adrenalectomy can be considered as an option if other therapies fail [122] [IIIC].
Clinically non-functioning pituitary tumours
• Pituitary surgery, usually by the trans-sphenoidal approach, is the treatment of choice [122] [IVA]
• Radiotherapy is indicated for recurrent or partially removed tumours [122] [IVA]
Von Hippel–Lindau syndrome: clinical and molecular diagnosis
A germline mutation in the short arm of chromosome 3 causes the inhibition of von Hippel–Lindau protein, which results in the increased hypoxia-inducing factor leading to increased production of PDGF, a stimulator of epithelial and vascular cell growth, that promotes hypervascular cystic tumours, carcinomas including clear renal cells and pancreatic NET, with a penetrance of approximately 15%, as well as paragangliomas and pheochromocytomas [126].
Recommendations
• All patients with a clinical diagnosis of VHL syndrome, even without a family history, should carry out a molecular test [126] [IVA].
• We recommend screening VHL patients with usual clinical evaluation and annual imaging tests (abdominal US, CT or MRI) beginning at 20 years of age [VB]. To differentiate pancreatic cystadenoma and cysts with septate and thick content, we suggest octreoscans or PET/CT68Ga, if available.
• Genetic testing is also recommended for the first-degree and second-degree family members to identify asymptomatic carriers of VHL disease [VA].
VHL: management of localised gastrointestinal disease
Recommendations
• We recommend radiological monitoring in cases of cysts in the pancreas. Surgery is indicated in exceptional cases for the compression of adjacent organs or development of NET.
• We recommend the approach used by Libutti et al [126] (Table 4) for patients with NETs of the pancreas [IVB].
Distinct management for metastatic NETs associated with hereditary syndromes
Therapeutic management of GEP NET associated with hereditary syndromes should follow the same principles of sporadic NET. Only case reports are available to support management of these tumours.
Recommendations
• In general, we recommend the use of sporadic tumour treatment protocols [VA].
• We prefer sunitinib as the first choice in pancreatic NET associated with VHL syndrome [VC] and everolimus as the first choice in MEN-1-associated pancreatic NET [119][VC]
Conclusion
The management of NETs is challenging and complex because of their vast heterogeneity. Thus, each case should be discussed and handled in a multidisciplinary environment. The development of national guidelines is an important roadmap for assertive diagnostic and therapeutic choices, not only based on scientific evidences but also adaptable to particular scenarios. In this Brazilian NET guideline, we constructed recommendations for diagnosis and treatment that differ from those found in current guidelines. We believe that our considerations are useful and can assist medical decisions for the best management of patients.
The incidence of NETs, although still low, has increased and will become more frequent in medical practice. Continuous efforts toward updated knowledge should be made, especially through collaborative groups. Working together can be more effective in standardising procedures, evaluating risks and benefits of available treatments, and optimising resources. Finally, clinical and translational research should be encouraged as it is essential in elucidating the carcinogenesis mechanisms of NETs and a key step in therapeutic advances.
Table 4: Management of Pancreatic NET in von Hippel–Lindal Syndrome.
References
1. Bosman FT (2010) WHO Classification of Tumours of the Digestive System (Lyon, France: IARC Press)
2. Klimstra DS (2013) Pathology reporting of neuroendocrine tumours: essential elements for accurate diagnosis, classification, and staging Semin Oncol 40 23–36 DOI: 10.1053/j.seminoncol.2012.11.001 PMID: 23391110
3. Klimstra DS et al (2010) Pathology reporting of neuroendocrine tumours: application of the Delphic consensus process to the development of a minimum pathology data set Am J Surg Pathol 34 300–313 DOI: 10.1097/PAS.0b013e3181ce1447 PMID: 20118772
4. Luo G et al (2016) Modified staging classification for pancreatic neuroendocrine tumours on the basis of the American Joint Committee on Cancer and European Neuroendocrine Tumour Society systems J Clin Oncol 34 [ahead of print]
5. Klimstra DS et al (2010) The pathologic classification of neuroendocrine tumours: a review of nomenclature, grading, and staging systems Pancreas 39 707–712 DOI: 10.1097/MPA.0b013e3181ec124e PMID: 20664470
6. Lawrence B et al (2011) The epidemiology of GEP neuroendocrine tumours Endocrinol Metab Clin North Am 40 1–18 DOI: 10.1016/j.ecl.2010.12.005
7. Massironi S et al (2014) Chromogranin A in diagnosing and monitoring patients with GEP neuroendocrine neoplasms: a large series from a single institution Neuroendocrinology 100 240–249 DOI: 10.1159/000369818
8. Rossi RE et al (2015) Chromogranin A as a predictor of radiological disease progression in neuroendocrine tumours Ann Transl Med 9 118
9. Turner GB et al (2006) Circulating markers of prognosis and response to treatment in patients with midgut carcinoid tumours Gut 55 1586–1591 DOI:
10. Okabayashi T et al (2013) Diagnosis and management of insulinoma World J Gastroenterol 19 829 DOI: 10.3748/wjg.v19.i6.829 PMID: 23430217 PMCID: 3574879
11. Cryer PE et al (2009) Evaluation and management of adult hypoglycemic disorders: an Endocrine Society Clinical Practice Guideline J Clin Endocrinol Metab 3 709–728 DOI: 10.1210/jc.2008-1410
12. Guettier J-M et al (2013) The role of proinsulin and insulin in the diagnosis of insulinoma: a critical evaluation of the endocrine society clinical practice guideline J Clin Endocrinol Metab 12 4752–4758 DOI: 10.1210/jc.2013-2182
13. Hirshberg B et al (2000) Forty-eight-hour fast: the diagnostic test for insulinoma J Clin Endocrinol Metab 9 3222–3226 DOI: 10.1210/jcem.85.9.6807
14. Kulke MH et al (2010) NANETS treatment guidelines: well-differentiated neuroendocrine tumours of the stomach and pancreas Pancreas 39 735–752 DOI: 10.1097/MPA.0b013e3181ebb168 PMID: 20664472 PMCID: 3100728
15. Berna MJ et al (2006) Serum gastrin in Zollinger–Ellison syndrome: II. Prospective study of gastrin provocative testing in 293 patients from the National Institutes of Health and comparison with 537 cases from the literature. evaluation of diagnostic criteria, proposal of new criteria, and correlations with clinical and tumoural features Medicine (Baltimore) 85 331–364 DOI: 10.1097/MD.0b013e31802b518c
16. Sundin A et al (2009) ENETS consensus guidelines for the standards of care in neuroendocrine tumours: radiological examinations Neuroendocrinology 90 167–183 DOI: 10.1159/000184855
17. Anderson MA et al (2000) Endoscopic ultrasound is highly accurate and directs management in patients with neuroendocrine tumours of the pancreas Am J Gastroenterol 95 2271–2277 DOI: 10.1111/j.1572-0241.2000.02480.x PMID: 11007228
18. Gouya H et al (2003) CT, endoscopic sonography, and a combined protocol for preoperative evaluation of pancreatic insulinomas AJR Am J Roentgenol 181 987–989 DOI: 10.2214/ajr.181.4.1810987 PMID: 14500214
19. Khashab MA et al (2011) EUS is still superior to multidetector computerized tomography for detection of pancreatic neuroendocrine tumours Gastrointest Endosc 73 691–696 DOI: 10.1016/j.gie.2010.08.030
20. Horton KM et al (2006) Multi-detector row CT of pancreatic islet cell tumours Radiographics 2 453–464 DOI: 10.1148/rg.262055056
21. Versari A et al (2010) Ga-68 DOTATOC PET, endoscopic ultrasonography, and multidetector CT in the diagnosis of duodenopancreatic neuroendocrine tumours: a single-centre retrospective study Clin Nucl Med 35 321–328 DOI: 10.1097/RLU.0b013e3181d6677c PMID: 20395703
22. Caramella C et al (2010) Endocrine pancreatic tumours: which are the most useful MRI sequences? Eur Radiol 11 2618–2627 DOI: 10.1007/s00330-010-1840-5
23. Baumann T et al (2016) Gastroenteropancreatic neuroendocrine tumours (GEP-NET) - Imaging and staging Best Pract Res Clin Endocrinol Metab 30 45–57 DOI: 10.1016/j.beem.2016.01.003 PMID: 26971843
24. Fidler JL et al (2017) Small bowel imaging: computed tomography enterography, magnetic resonance enterography, angiography, and nuclear medicine Gastrointest Endosc Clin N Am 27 133–152 DOI: 10.1016/j.giec.2016.08.008
25. Puli SR et al (2013) Diagnostic accuracy of endoscopic ultrasound in pancreatic neuroendocrine tumours: a systematic review and meta-analysis World J Gastroenterol 23 3678–3684 DOI: 10.3748/wjg.v19.i23.3678
26. Sugimoto M et al (2015) Conventional versus contrast-enhanced harmonic endoscopic ultrasonography-guided fine-needle aspiration for diagnosis of solid pancreatic lesions: a prospective randomized trial Pancreatology 15 538–541 DOI: 10.1016/j.pan.2015.06.005 PMID: 26145837
27. Reidy-Lagunes D et al (2011) Assition of octreotide functional imaging to cress-sectional computed tomography or magnetic resonance imaging for the detection of neuroendocrine tumours: added value or an anachronism? J Clin Oncol 20 e74–e75 DOI: 10.1200/JCO.2010.32.8559
28. Geijer H and Breimer LH (2013) Somatostatin receptor PET/CT in neuroendocrine tumours: update on systematic review and meta-analysis Eur J Nucl Med Mol Imaging 11 1770–1780 DOI: 10.1007/s00259-013-2482-z
29. Etchebehere EC et al (2014) 68Ga-DOTATATE PET/CT, 99mTc-HYNIC-octreotide SPECT/CT, and whole-body MR imaging in detection of neuroendocrine tumours: a prospective trial J Nucl Med 10 1598–1604 DOI: 10.2967/jnumed.114.144543
30. Panagiotidis E et al (2016) Comparison of the impact of 68Ga – DOTATATE and 18F – DG PET/CT on clinical management in patients with neuroendocrine tumours J Nucl Med DOI: 10.2967/jnumed.116.178095
31. Panagiotidis E and Bomanji J (2016) Role of 18F-fluorodeoxyglucose PET in the study of neuroendocrine tumours PET Clin 9 43–55 DOI: 10.1016/j.cpet.2013.08.008
32. Naswa N et al (2012) Ga-DOTANOC PET/CT in patients with carcinoma of unknown primary of neuroendocrine origin Clin Nucl Med 37 245–251 DOI: 10.1097/RLU.0b013e31823ea730 PMID: 22310250</a>
33. Nakamoto Y et al (2015) Additional information gained by positron emission tomography with (68)Ga-DOTATOC for suspected unknown primary or recurrent neuroendocrine tumours Ann Nucl Med 29 512–518 DOI: 10.1007/s12149-015-0973-7 PMID: 25894056 PMCID: 4661205
34. Moryoussef F et al (2016) Impact on management of liver and whole-body diffusion weighted magnetic resonance imaging sequences for neuroendocrine tumours: a pilot study Neuroendocrinology 28. [Epub ahead of print]
35. Boudreaux JP et al (2005) Surgical treatment of advanced-stage carcinoid tumours: lessons learned Ann Surg 241 839–845 DOI: 10.1097/01.sla.0000164073.08093.5d
36. Hellman P et al (2002) Effect of surgery on the outcome of midgut carcinoid disease with lymph node and liver metastases World J Surg 8 991–997 DOI: 10.1007/s00268-002-6630-z
37. Givi B et al (2006) Operative resection of primary carcinoid neoplasms in patients with liver metastases yields significantly better survival Surgery 140 891–897 DOI: 10.1016/j.surg.2006.07.033 PMID: 17188135
38. Bartlett EK et al (2013) Surgery for metastatic neuroendocrine tumours with occult primaries J Surg Res 184 221–227 DOI: 10.1016/j.jss.2013.04.008 PMID: 23643298
39. Bellutti M (2009) Detection of neuroendocrine tumours of the small bowel by double balloon enteroscopy Dig Dis Sci 54 1050–1058 DOI: 10.1007/s10620-008-0456-y
40. Naswa N et al (2012) 68Ga-DOTANOC PET/CT in patients with carcinoma of unknown primary of neuroendocrine origin Clin Nucl Med 37 245–251 DOI: 10.1097/RLU.0b013e31823ea730 PMID: 22310250
41. Prasad V et al (2010) Detection of unknown primary neuroendocrine tumours (CUP-NET) using 68 Ga-DOTA-NOC receptor PET/CT Eur J Nucl Med Mol Imaging 37 67–77 DOI: 10.1007/s00259-009-1205-y
42. Kunikowska J et al (2014) A frequency and semiquantitative analysis of pathological 68Ga DOTATATE PET/CT uptake by primary site-dependent neuroendocrine tumour metastasis Clin Nucl Med 39 855–861 DOI: 10.1097/RLU.0000000000000533 PMID: 25072928
43. Tan et al (2014) Diagnostic value of (68)Ga-DOTATATE PET/CT in liver metastases of neuroendocrine tumours of unknown origin Nucl Med Mol Imaging 48 212–215 DOI: 10.1007/s13139-013-0258-9 PMID: 25177378 PMCID: 4145102
44. Savelli G et al (2004) Feasibility of somatostatin receptor scintigraphy in the detection of occult primary gastro-entero-pancreatic (GEP) neuroendocrine tumours Nucl Med Commun 25 445–449 DOI: 10.1097/00006231-200405000-00004 PMID: 15100502
45. Chang S et al (2007) Neuroendocrine neoplasms of the gastrointestinal tract: classification, pathologic basis and imaging features Radiographics 27 1667–1679 DOI: 10.1148/rg.276075001 PMID: 18025510
46. Van Tuyl et al (2006) Detection of small-bowel neuroendocrine tumours by video capsule endoscopy Gastrointest Endosc 64 66–72 DOI: 10.1016/j.gie.2006.01.054 PMID: 16813805
47. Frilling A et al (2014) Capsule endoscopy to detect primary tumour site in metastatic neuroendocrine tumours Dig Liver Dis 46 1038–1042 DOI: 10.1016/j.dld.2014.07.004 PMID: 25086997
48. Karagiannis S et al (2009) Capsule retention: a feared complication of wireless capsule endoscopy Scand J Gastroenterol 44 1158–1165 DOI: 10.1080/00365520903132039 PMID: 19606392
49. Oberg K and Castellano D (2011) Current knowledge on diagnosis and staging of neuroendrocrine tumours Cancer Metastasis Rev 30(Suppl 1) 3–7 DOI: 10.1007/s10555-011-9292-1
50. Syngal S et al (2005) Surveillance of patients at high risk for colorectal cancer Med Clin North Am 89 61–84 DOI: 10.1016/j.mcna.2004.08.013
51. Herrera MF et al (2015) AACE/ACE disease state clinical review: pancreatic neuroendocrine incidentalomas Endocr Pract 21 546–553 DOI: 10.4158/EP14465.DSC PMID: 25962093 PMCID: 4975928
52. Jung JG et al (2015) Behaviour of small, asymptomatic, nonfunctioning pancreatic neuroendocrine tumours (NF-PNETs) Medicine (Baltimore) 94 e983 DOI: 10.1097/MD.0000000000000983
53. Partelli S et al (2016) Systematic review of active surveillance versus surgical management of asymptomatic small non-functioning pancreatic neuroendocrine neoplasm Br J Surg
54. Capurso G et al (2012) Systematic review of resection of primary midgut carcinoid tumour in patients with unresectable liver metastases Br J Surg 99 1480–1486 DOI: 10.1002/bjs.8842 PMID: 22972490
55. Keutgen XM et al (2016) Non-functioning pancreatic endocrine tumour Surgery 159 311–318 DOI: 10.1016/j.surg.2015.05.042
56. Bettini R et al (2009) Primary tumour resection in metastatic nonfunctioning pancreatic endocrine carcinomas Dig Liver Dis 41 49–55 DOI: 10.1016/j.dld.2008.03.015
57. Charnsangavej C et al (2006) Selection of patients for resection of hepatic colorectal metastases: expert consensus statement Ann Surg Oncol 10 1261–1268 DOI: 10.1245/s10434-006-9023-y
58. Abdalla EK et al (2013) Locoregional surgical and interventional therapies for advanced colorectal cancer liver metastases: expert consensus statements HPB (Oxford) 15 119–130 DOI: 10.1111/j.1477-2574.2012.00597.x
59. Kianmanesh R et al (2015) Surgical treatment of gastric, enteric, and pancreatic endocrine tumours Part 1. Treatment of primary endocrine tumours J Chir 142 132–149 DOI: 10.1016/S0021-7697(05)80881-6
60. Sarmiento JM et al (2003) Surgical treatment of neuroendocrine metastases to the liver: a plea for resection to increase survival J Am Coll Surg 197 29–37 DOI: 10.1016/S1072-7515(03)00230-8 PMID: 12831921
61. Chen J et al (2011) Simultaneous vs. staged resection for synchronous colorectal liver metastases: a metaanalysis Int J Colorectal Dis 2 191–199 DOI: 10.1007/s00384-010-1018-2
62. Graff-Baker AN et al (2014) Expanded criteria for carcinoid liver debulking: maintaining survival and increasing the number of eligible patients Surgery 156 1369–1376 DOI: 10.1016/j.surg.2014.08.009 PMID: 25456912
63. Hodul P et al (2006) The role of cytoreductive hepatic surgery as an adjunct to the management of metastatic neuroendocrine carcinomas Cancer Control 1 61–71
64. D’Haese JG et al (2014) Update on surgical treatment of pancreatic neuroendocrine neoplasm World J Gastroenterol 38 13893–13898 DOI: 10.3748/wjg.v20.i38.13893
65. Kennedy A et al (2015) Role of hepatic intra-arterial therapies in metastatic neuroendocrine tumours (NET): guidelines from the NET-Liver-Metastases Consensus Conference HPB (Oxford) 17 29–37 DOI: 10.1111/hpb.12326
66. Frilling A and Clift AK (2015) Therapeutic strategies of neuroendocrine liver metastases Cancer 121 1172–1186 DOI: 10.1002/cncr.28760
67. Lewis M and Hobday T (2012) Treatment of neuroendocrine tumour liver metastases Int J Hepatol 1–12 DOI: 10.1155/2012/973946
68. Coldwell (2013) Treatment of NET with SIRT Liver Radioembolization with Y90 Microspheres ed JI Bilbao and MF Reiser (Berlin: Springer) p 153
69. Device Z et al (2014) The efficacy of hepatic 90Y resin radioembolization for metastatic neuroendocrine tumours: a meta-analysis J Nucl Med 55 1404–1410 DOI: 10.2967/jnumed.113.135855
70. Yang TX et al (2012) Radioembolization and chemoembolization for unresectable neuroendocrine liver metastases - a systematic review Surg Oncol 21 299–308 DOI: 10.1016/j.suronc.2012.07.001 PMID: 22846894
71. Mazzaferro V et al (2007) Neuroendocrine tumours metastatic to the liver: how to select patients for liver transplantation? J Hepatol 47 460–466 DOI: 10.1016/j.jhep.2007.07.004 PMID: 17697723
72. Mazzaferro V et al (1996) Liver transplantation for the treatment of small hepatocellular carcinomas in patients with cirrhosis N Engl J Med 334 693–699 DOI: 10.1056/NEJM199603143341104 PMID: 8594428
73. Rossi RE et al (2014) Liver transplantation for unresectable neuroendocrine tumour liver metastases Ann Surg Oncol 7 2398–2405 DOI: 10.1245/s10434-014-3523-y
74. Pape UF et al (2012) ENETS Consensus Guidelines for the management of patients with neuroendocrine neoplasms from the jejuno-ileum and the appendix including goblet cell carcinomas Neuroendocrinology 95 135–156 DOI: 10.1159/000335629 PMID: 22262080
75. Caplin ME et al (2014) Lanreotide in metastatic enteropancreatic neuroendocrine tumours N Engl J Med 371 224–233 DOI: 10.1056/NEJMoa1316158 PMID: 25014687
76. Rinke A et al (2009) Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumour growth in patients with metastatic neuroendocrine midgut tumours: a report from the PROMID Study Group J Clin Oncol 27 4656–4663 DOI: 10.1200/JCO.2009.22.8510 PMID: 19704057
77. Janson ET et al (1994) [111in-DTPA-D-Phe1] octreotide scintigraphy in patients with carcinoid tumours: the predictive value for somatostatin analogue treatment Eur J Endocrinol 131 577–581 DOI: 10.1530/eje.0.1310577 PMID: 7804439
78. Riechelmann R et al (2016) Refractory carcinoid syndrome: a review of treatment options Ther Adv Med Oncol 1–11
79. Strosberg J et al (2015) The Role of chemotherapy in well-differentiated GEP neuroendocrine tumours Front Horm Res 44 239–247 DOI: 10.1159/000403785
80. Ferrarotto R et al (2013) Combination of capecitabine and oxaliplatin is an effective treatment option for advanced neuroendocrine tumours Rare Tumours 24 e35 DOI: 10.4081/rt.2013.e35
81. Dussol AS et al (2015) Gemcitabine and oxaliplatin or alkylating agents for neuroendocrine tumours: comparison of efficacy and search for predictive factors guiding treatment choice Cancer 121 3428–3434 DOI: 10.1002/cncr.29517 PMID: 26058464
82. Pavel ME et al (2011) Everolimus plus octreotide long-acting repeatable for the treatment of advanced neuroendocrine tumours associated with carcinoid syndrome (RADIANT-2): a randomised, placebo-controlled, phase 3 study Lancet 378 2005–2012 DOI: 10.1016/S0140-6736(11)61742-X PMID: 22119496
83. Yao JC et al (2016) Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (RADIANT-4): a randomised, placebo-controlled, phase 3 study Lancet 387 968–977 DOI: 10.1016/S0140-6736(15)00817-X
84. Kulke MH et al (2009) Glycemic control in patients with insulinoma treated with everolimus N Engl J Med 360 195–197 DOI: 10.1056/NEJMc0806740 PMID: 19129539
85. Kulke MH et al (2008) Activity of sunitinib in patients with advanced neuroendocrine tumours J Clin Oncol 26 3403–3410 DOI: 10.1200/JCO.2007.15.9020 PMID: 18612155
86. Yao JC et al (2011) Everolimus for advanced pancreatic neuroendocrine tumours N Engl J Med 364 514–523 DOI: 10.1056/NEJMoa1009290 PMID: 21306238 PMCID: 4208619
87. Raymond E et al (2011) Sunitinib malate for the treatment of pancreatic neuroendocrine tumours N Engl J Med 364 501–513 DOI: 10.1056/NEJMoa1003825 PMID: 21306237
88. Iacovelli R et al (2012) Incidence and risk of pulmonary toxicity in patients treated with mTOR inhibitors for malignancy. A meta-analysis of published trials Acta Oncol 7 873–879 DOI: 10.3109/0284186X.2012.705019
89. Bodei L et al (2013) The joint IAEA, EANM, and SNMMI practical guidance on peptide receptor radionuclide therapy (PRRNT) in neuroendocrine tumours Eur J Nucl Med Mol Imaging 40 800–816 DOI: 10.1007/s00259-012-2330-6 PMID: 23389427 PMCID: 3622744
90. Kwekkeboom DJ et al (2008) Treatment with the radiolabeled somatostatin analogue [177 Lu-DOTA 0,Tyr3] octreotate: toxicity, efficacy, and survival J Clin Oncol 26 2124–2130 DOI: 10.1200/JCO.2007.15.2553 PMID: 18445841
91. Strosberg JR et al (2017) Netter 1 phase III: progression free-survival, radiographic response, and preliminary overall survival results in patients with midgut neuroendocrine tumours treated with 177 lu dotate N Engl J Med 376(2) 125
92. Leboulleux S et al (2008) Prediction and diagnosis of metastases in well-differentiated gastro-entero-pancreatic endocrine cancer: a prospective comparison of whole body magnetic resonance imaging and somatostatin receptor scintigraphy J Clin Endocrinol Metab 93 3021–3028 DOI: 10.1210/jc.2008-0459 PMID: 18522978
93. Lebtahi R et al (1999) Detection of bone metastases in patients with endocrine GEP tumours: bone scintigraphy compared with somatostatin receptor scintigraphy J Nucl Med 40 1602–1608 PMID: 10520698
94. Janson ET et al (1997) Carcinoid tumours: analysis of prognostic factors and survival in 301 patients from a referral center Ann Oncol 8 685–690 DOI: 10.1023/A:1008215730767 PMID: 9296223
95. Van Loon K et al (2015) Bone metastases and skeletal-related events from neuroendocrine tumours Endocr Connect 4 9–17 DOI: 10.1530/EC-14-0119 PMCID: 4285767
96. Van Binnebeek S et al (2016) Comparison of diagnostic accuracy of (111)In-pentetreotide SPECT and (68)Ga-DOTATOC PET/CT: a lesion-by-lesion analysis in patients with metastatic neuroendocrine tumours Eur Radiol 26 900–909 DOI: 10.1007/s00330-015-3882-1
97. Buchmann I et al (2007) Comparison of 68Ga-DOTATOC PET and 111In-DTPAOC (Octreoscan) SPECT in patients with neuroendocrine tumours Eur J Nucl Med Mol Imaging 34 1617–1626 DOI: 10.1007/s00259-007-0450-1 PMID: 17520251
98. Mota JM et al (2016) Complications from carcinoid syndrome: review of the current evidence Ecancermedicalscience 8 662
99. Kulke MH et al (2016) Telotristat ethyl, a tryptophan hydroxylase inhibitor for the treatment of carcinoid syndrome J Clin Oncol 28
100. Moerman VM et al (2012) Carcinoid heart disease: typical findings on echocardiography and cardiac magnetic resonance Acta Cardiol 67 245–248 PMID: 22641985
101. Connolly HM et al (2015) Early and late outcomes of surgical treatment in carcinoid heart disease J Am Coll Cardiol 66 2189–2196 DOI: 10.1016/j.jacc.2015.09.014 PMID: 26564596
102. Jensen RT et al (2006) Gastrinoma (duodenal and pancreatic) Neuroendocrinology 84 173–182 DOI: 10.1159/000098009
103. Stephen AE and Hodin RA (2006) Neuroendocrine tumours of the pancreas, excluding gastrinoma Surg Oncol Clin N Am 15 497–510 DOI: 10.1016/j.soc.2006.05.012 PMID: 16882494
104. Munhoz RR et al (2013) Combination of irinotecan and a platinum agent for poorly differentiated neuroendocrine carcinomas Rare Tumours 5 e39 DOI: 10.4081/rt.2013.e39
105. Bezerra EB et al (2011) Poorly differentiated neuroendocrine tumours Neuroendocrine Tumour 157–176 DOI: 10.1007/978-1-60327-997-0_10
106. Sorbye H et al (2014) GEP high-grade neuroendocrine carcinoma Cancer 120 2814–2823 DOI: 10.1002/cncr.28721 PMID: 24771552
107. Tang LH et al (2016) A practical approach to the classification of WHO grade 3 (G3) well-differentiated neuroendocrine tumour (WD-NET) and poorly differentiated neuroendocrine carcinoma (PD-NEC) of the pancreas Am J Surg Pathol 40 1192–1202 DOI: 10.1097/PAS.0000000000000662 PMID: 27259015 PMCID: 4988129
108. Walter DT et al (2015) Characteristics, prognosis and treatments of 294 patients with poorly differentiated neuroendocrine carcinoma: the FFCD-GTE national cohort J Clin Oncol 33(May 20 Supplement) 4095
109. Welin S et al (2011) Clinical effect of temozolomide-based chemotherapy in poorly differentiated endocrine carcinoma after progression on first line chemotherapy Cancer 117 4617–4622 DOI: 10.1002/cncr.26124 PMID: 21456005
110. Marx SJ (2005) Molecular genetics of multiple endocrine neoplasia types 1 and 2 Nat Rev Cancer 5 367–375 DOI: 10.1038/nrc1610 PMID: 15864278
111. Brandi ML et al (2001) Guidelines for diagnosis and therapy of MEN type 1 and type 2 J Clin Endocrinol Metab 86 5658–5671 DOI: 10.1210/jcem.86.12.8070 PMID: 11739416
112. Lourenço DM Jr et al (2007) The impact of clinical and genetic screenings on the management of the multiple endocrine neoplasia type 1 Clinics 62 465–476 DOI: 10.1590/S1807-59322007000400014
113. Thakker RV et al (2012) Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1) J Clin Endocrinol Metab 97 2990–3011 DOI: 10.1210/jc.2012-1230 PMID: 22723327
114. Norton JA et al (2006) Surgery increases survival in patients with gastrinoma Ann Surg 244 410–419 PMID: 16926567 PMCID: 1856542
115. Bartsch DK et al (2005) Outcome of duodenopancreatic resections in patients with multiple endocrine neoplasia type 1 Ann Surg 242 757–764 DOI: 10.1097/01.sla.0000189549.51913.d8
116. Jensen RT et al (2012) ENETS Consensus Guidelines for the management of patients with digestive neuroendocrine neoplasms: functional pancreatic endocrine tumour syndromes Neuroendocrinology 95 98–119 DOI: 10.1159/000335591
117. Triponez F et al (2006) Epidemiology data on 108 MEN 1 patients from the GTE with isolated nonfunctioning tumours of the pancreas Ann Surg 243 265–272 DOI: 10.1097/01.sla.0000197715.96762.68 PMID: 16432361 PMCID: 1448903
118. Gonçalves TD et al (2014) Penetrance of functioning and nonfunctioning pancreatic neuroendocrine tumours in multiple endocrine neoplasia type 1 in the second decade of life J Clin Endocrinol Metab 99 e89–96 DOI: 10.1210/jc.2013-1768
119. Maia MC et al (2016) Efficacy and long-term safety of everolimus in pancreatic neuroendocrine tumour associated with multiple endocrine neoplasia type I: case report Oncol Res Treat 39 643–645 DOI: 10.1159/000448699
120. Webster J et al (1994) A comparison of cabergoline and bromocriptine in the treatment of hyperprolactinemic amenorrhea. Cabergoline Comparative Study Group N Engl J Med 331 904–909 DOI: 10.1056/NEJM199410063311403 PMID: 7915824
121. Glezer A and Bronstein MD (2015) Prolactinomas Endocrinol Metab Clin North Am 44 71–78 DOI: 10.1016/j.ecl.2014.11.003 PMID: 25732643
122. Giustina A et al (2014) Expert consensus document: a consensus on the medical treatment of acromegaly Nat Rev Endocrinol 10 243–248. DOI: 10.1038/nrendo.2014.21 PMID: 24566817
123. Trainer PJ et al (2009) A randomized, controlled, multicentre trial comparing pegvisomant alone with combination therapy of pegvisomant and long-acting octreotide in patients with acromegaly Clin Endocrinol (Oxf) 71 549–557 DOI: 10.1111/j.1365-2265.2009.03620.x
124. Kasuki L et al (2016) Experience with pegvisomant treatment in acromegaly in a single Brazilian tertiary reference center: efficacy, safety and predictors of response Arch Endocrinol Metab 60 479–485 DOI: 10.1590/2359-3997000000210 PMID: 27737325
125. McCormack AI et al (2011) Aggressive pituitary tumours: the role of temozolomide and the assessment of MGMT status Eur J Clin Invest 41 1133–1148 DOI: 10.1111/j.1365-2362.2011.02520.x PMID: 21496012
126. Libutti SK et al (2000) Clinical and genetic analysis of patients with pancreatic neuroendocrine tumours associated with von Hippel–Lindau disease Surgery 128 1022–1027 DOI: 10.1067/msy.2000.110239
127. Riechelmann R et al (2016) Guideline for the management of bile duct cancers by the Brazilian gastrointestinal tumour group Arq Gastroenterol 53 5–9 DOI: 10.1590/S0004-28032016000100003 PMID: 27276097
128. Libutti SK et al (1998) Pancreatic neuroendocrine tumours associated with von Hippel–Lindau disease: diagnostic and management recommendations Surgery 124 1153–1159 DOI: 10.1067/msy.1998.91823 PMID: 9854597





