The development of T-cell malignancies in patients with pre-existing myeloproliferative neoplasms: a report of three cases
Ethan A Burns1,*, Kartik Anand2,*, Betty Chung3, Shilpan Shah2 Jasleen K Randhawa2 and Sai Ravi Pingali2
1Department of Internal Medicine, Houston Methodist Hospital, 6550 Fannin, Smith Tower, Ste 1101, Houston, TX 77030, USA
2Houston Methodist Cancer Center, 6445 Main Street, Outpatient Center, 24th Floor, Houston, TX 77030, USA
3Houston Methodist Hospital, Department of Pathology and Genomic Medicine, 6550 Fannin St, Houston, TX 77030, USA
*Equal contribution.
Abstract
Secondary acute myeloid leukaemia complicating the natural disease course of pre-existing Philadelphia chromosome-negative myeloproliferative neoplasms (PN-MPN) is well documented and associated with treatment challenges and significant morbidity. The incidence of a T-cell malignancy developing in patients with pre-existing PN-MPN is uncommon, with one case documented in the literature. We present two cases of angioimmunoblastic T-cell lymphoma (AITL) and one case of T-cell acute lymphoblastic leukaemia (T-ALL) that developed in patients with essential thrombocythemia (ET) and primary myelofibrosis (PMF), respectively. All malignancies were advanced at diagnosis and exhibited disease progression, regardless of the mutational status of the underlying ET/PMF, presence of cytogenetic abnormalities, type of T-cell neoplasm or systemic chemotherapy utilised. The median time to diagnosis of AITL or T-ALL from the onset of MPN was 4.5 years (range: 6 months–10 years). This single institutional case series demonstrates the possibility of an association between T-cell neoplasms and PN-MPNs.
Keywords: essential thrombocythemia, primary myelofibrosis, polycythemia vera, Philadelphia negative myeloproliferative neoplasm, angioimmunoblastic T-cell lymphoma, T-cell acute lymphoblastic leukaemia
Correspondence to: Ethan A Burns
Email: Eaburns@houstonmethodist.org
Published: 17/02/2020
Received: 12/09/2019
Publication costs for this article were supported by ecancer (UK Charity number 1176307).
Copyright: © the authors; licensee ecancermedicalscience. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Introduction
Myeloproliferative neoplasms (MPNs) are chronic, clonal diseases defined by the overproduction of fully differentiated cells of haematopoietic origin. In 2016, the World Health Organization Revised Classification System for Haematopoietic Tumours recognised a subcategory of Philadelphia chromosome (PN)-MPN, including polycythemia vera (PV), essential thrombocythemia (ET), primary myelofibrosis (PMF) and prefibrotic PMF [1, 2]. PN-MPNs are phenotypically driven by somatic mutations which culminate in the persistent downstream hyperactivation of the Janus Kinase (JAK)-STAT (signal transducer and activator of transcription) signal transduction pathway. The most common mutations include JAK2 V617F, calreticulin (CALR) or the thrombopoietin receptor (MPL) [3]. Wild type (triple-negative) PMF or ET is not uncommon.
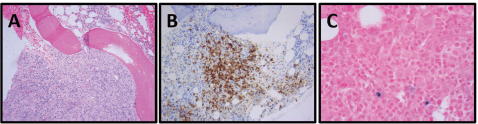
Figure 1. Patient 1, bone marrow biopsy with AITL: (A) Focal, interstitial marrow involvement with monomorphic population of small to medium-sized lymphoid cells with moderately abundant pale eosinophilic cytoplasm, H&E, 100×. (B) PD-1, 100×. (C) EBER ISH, 400×.
While there are similarities in the clinical, laboratory, morphologic and mutational manifestations of PN-MPNs, variations in survival and progression to leukaemic transformation necessitate an accurate diagnosis [1, 4]. The risk for leukaemic transformation in the decade following the diagnosis of a PN-MPN is 1.3% in ET [5], 2.3% in PV [6] and 10%–20% in PMF [7–9]. The lifetime cumulative incidence of a leukaemic transformation from MPN is approximately 3.8% for ET, 6.8% for PV and 14.2% for PMF [10]. While the most common haematologic malignancies following leukaemic transformation are of myeloid origin, lymphoid malignancies are seldom encountered [4]. The following three cases describe the development of T-cell neoplasms in patients with PN-MPN, a rarely reported occurrence.
Cases
Patient 1
A 78-year-old Caucasian woman with a history of pulmonary embolism and triple-negative ET diagnosed 6 months prior and treated with hydroxyurea presented to the hospital with shortness of breath and constipation.
She reported ‘B’ symptoms first noticed 1 month prior to admission. Physical examination was significant for axillary lymphadenopathy (LAD). Significant laboratory findings included hypercalcaemia of 13.6 mg/dL, acute kidney injury with admission creatinine of 2.03 mg/dL and pancytopenia. Computed Tomography (CT) showed splenomegaly (17 cm), diffuse lymphadenopathy and Positron Emission Tomography (PET) demonstrated increased uptake in the splenic and porta hepatis area. Her bone marrow biopsy found atypical lymphoid aggregates that contained a mixed population of T-cells positive for CD3, CD5 and PD1, and B-cells positive for CD20 and variably positive for PAX-5 and BCL6 (Figure 1; Table 1). Epstein Barr Virus encoded small ribonucleic acid (EBER) was highlighted in scattered, predominantly small lymphocytes. Karyotype analysis demonstrated multiple cytogenetic anomalies, with karyotype showing 47,XX,add(2)(p2),add(10)(p11.2), add(20)(p12)[cp4]/46,XX [16] (Table 1). Excisional axillary lymph node biopsy had immunostaining positive for CD2, CD3, CD5, CD7 and CD4 PD-1, and negative for CD30 in the lesional T-cells, consistent with AITL (Figure 2; Table 1).
She received three cycles of Cyclophosphamide, Doxorubicin Hydrochloride, Etoposide, Vincristine and Prednisone (CHEOP), but repeat PET/CT indicated disease progression in the spleen and increased uptake in her cervical, supraclavicular, axillary, hilar, mediastinal, inguinal and iliac lymph nodes. She was initiated on salvage Romidepsin, Ifosfamide, Carboplatin, Etoposide (R-ICE) and completed cycle 1, 5 months following the diagnosis of AITL and 11 months following the diagnosis of ET.
Patient 2
An 84-year-old Caucasian man with a history of congestive heart failure and ET (JAK2 V617F mutation) diagnosed 6 years prior and treated with hydroxyurea presented for evaluation of peripheral blood eosinophilia. He noted weight loss and fatigue but denied night sweats or fevers. Physical examination was notable for palpable cervical, axillary and inguinal LAD, and a diffuse macular rash. Initial laboratory findings were notable for a peripheral eosinophilia of 17.7%, peripheral monocytosis of 19.4% and lymphopenia of 12.4%. CT of his chest, abdomen and pelvis noted diffuse LAD and splenomegaly. His bone marrow biopsy showed hypercellular marrow (50%) without evidence of malignancy. Corresponding flow cytometry indicated an increased CD4:CD8 ratio of 10:1 without aberrant T-cell antigen expression or a clonal B-cell population. His axillary lymph node biopsy with immunostaining was positive for CD2, CD3, CD4, CD5, CD7, CD10 (subsets), BCL-6 and PD-1 in the lesional T-cells, consistent with AITL (Figure 3; Table 1). EBV was negative by EBER. Cytogenetic analysis demonstrated a normal male karyotype (46, XY).
His PET/CT showed increased uptake in the right cervical, infraparotid and axillary lymph nodes, right upper lobe of the lung, and spleen. He was started on Cyclophosphamide, Etoposide, Vincristine and Prednisone (CEOP); adriamycin hydrochloride was held due to his heart failure. After three cycles, repeat PET/CT showed disease progression in all involved sites. The patient ultimately requested comfort/hospice care, 4 months after the diagnosis of AITL and 6 years after the diagnosis of ET.
Table 1. Characteristics of patients with MPN with secondary AITL and T-ALL.
Patient 3
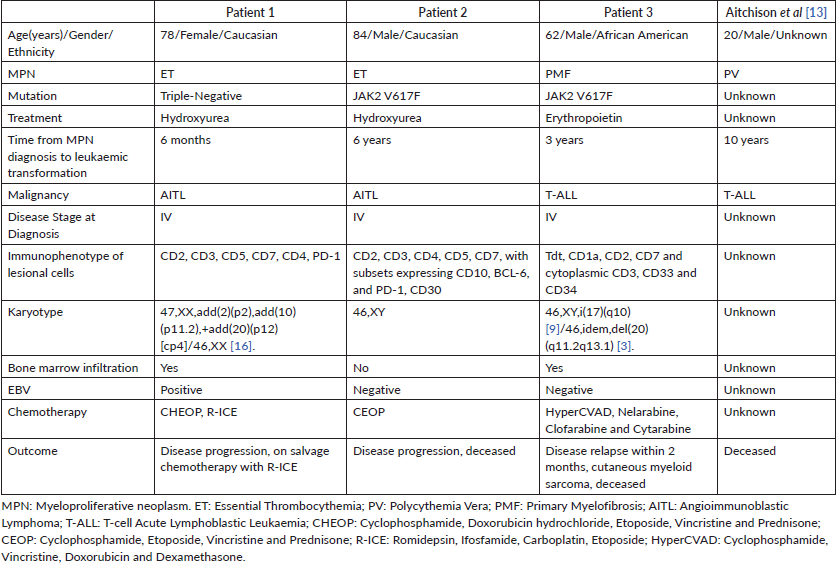
A 62-year-old African American man with a history of PMF (JAK2 V617F mutation) presented 3 years after initial diagnosis with a 30-pound weight loss, night sweats and fatigue. Physical examination revealed bilateral posterior cervical LAD. The patient had a bone marrow biopsy that indicated fibrosis with 40% cellularity, but no evidence of malignancy. Cytogenetic analysis showed 46,XY,i(17)(q10) [9]/46,idem,del(20)(q11.2q13.1) [3]. His excisional cervical lymph node biopsy immunophenotype stained positive for Tdt, CD1a, CD2, CD7, cytoplasmic CD3, CD33 and CD34, and was negative for surface CD3 and MPO in the lesional T-cells, compatible with the diagnosis of T-ALL (Figure 4; Table 1). His PET/CT showed increased uptake in the left cervical and axillary lymph nodes. He was initiated on Cyclophosphamide, Vincristine, Doxorubicin and Dexamethasone (HyperCVAD), and, after six cycles, was in complete remission.
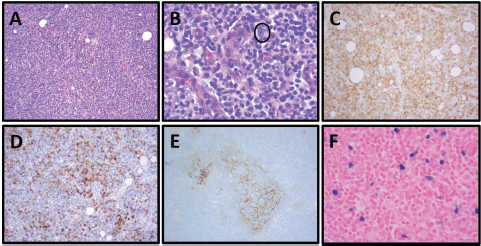
Figure 2. Patient 1, axillary lymph node with AITL: (A) Prominent vascular proliferation of high endothelial venules (HEVs) and nodal architecture effacement by a monomorphic lymphoid population, H&E, 100×. (B) Neoplastic population of follicular helper T cells with small to medium-sized nuclei and clear-to-pale cytoplasm, especially clustering adjacent to HEVs, and scattered CD30 immunoblasts (circled, IHC not shown). H&E, 400×. (C) Increased numbers of T cells in effaced areas, especially perivascular T cells, CD3, 100x. (D) Increased numbers of neoplastic follicular helper T cells, PD-1, 100×. (E) Expanded dendritic meshwork, CD21, 100×. (F) EBER in situ hybridisation (EBER ISH) positive neoplastic T cells, 100×.
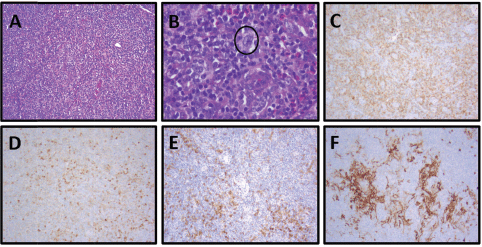
Figure 3. Patient 2, axillary lymph node with AITL: (A) Partial nodal architecture effacement by a monomorphic lymphoid population and increased numbers of eosinophils, H&E, 100×. (B) Neoplastic population of monomorphic follicular helper T cells with small to medium-sized nuclei and clear-to-pale cytoplasm in a polymorphous background of neutrophils and increased numbers of eosinophils with scattered CD30 immunoblasts (circled, IHC not shown), H&E, 400x. (C) Increased numbers of neoplastic helper T cells, CD4, 100×. (D) Increased numbers of neoplastic follicular T helper cells (dim) with scattered neutrophils (strong), CD10, 100x. (E) Increased numbers of neoplastic helper T cells, PD-1, 100x. (F) Expanded dendritic meshwork, CD23, 100×.
Two months later, he developed posterior cervical and supraclavicular LAD. His CBC was significant for pancytopenia, requiring transfusions of packed red blood cells. A repeat bone marrow biopsy indicated progressive marrow fibrosis and a focal area of TdT positivity. Bone marrow flow cytometry was negative for lymphoblastic cells. The patient had a CT scan of his chest, abdomen and pelvis, which demonstrated splenomegaly (18 cm) and diffuse LAD. A core biopsy of a left cervical lymph node was consistent with relapsed T-ALL (stage IV), so the patient was started on Nelarabine and Dexamethasone. After two cycles, the patient developed a diffuse papular rash. He had a punch biopsy that showed an atypical immature cell infiltrate within the dermis. Immunochemical staining was positive for CD45, MPO, CD43 and CD68, and negative for CD3, CD5, TdT, CD20, CD34, CD117 and CD1a in the lesional cells, consistent with the diagnosis of a cutaneous myeloid sarcoma. He was transitioned to Clofarabine and Cytarabine, which was complicated by neutropenic sepsis from Escherichia Coli bacteremia and acute appendicitis after cycle 1. Ultimately, the patient requested comfort/hospice care, 3 years after being diagnosed with PMF, and 1 year following the diagnosis of T-ALL.
Discussion
These cases demonstrate the development of AITL and T-ALL in patients previously diagnosed with PN-MPN. This is the first case series of T-ALL or AITL arising in patients with PN-MPN, apart from one other reported case in the literature [11] (Table 1). While rarely described, these cases suggest a possible association between PN-MPN and T-cell malignancies. While the development of secondary lymphoid neoplasms remains rare, data suggest that the incidence is greater in males, in those with a JAK2 V617F mutation, and in patients with a pre-existing PN-MPN of greater than 5 years [12]. When including the other reported case with this case series, the median age was 70 years (range: 20–84 years) with a 3:1 male-to-female predominance, and a median duration of 4.5 years (range: 6 months–10 years) between the diagnosis of a PN-MPN and the development of a T-cell neoplasm (Table 1). All cases were advanced at diagnosis and exhibited rapid progression or relapse despite systemic chemotherapy and regardless of karyotype, MPN mutation and type of PN-MPN or T-cell neoplasm (Table 1).
The occurrence of a haematolymphoid malignancy following a previously diagnosed PN-MPN is uncommon. Nonetheless, clinicians should be aware that studies have demonstrated patients with PN-MPN are at increased risk of this complication. Vannucchi et al [12] reported a standardised incidence ratio (SIR) of 3.4 (95% confidence interval (95% CI): 1.9–6.2) and 12.4 (95% CI: 4.7–33.6) in patients who develop secondary chronic lymphocytic leukaemia and non-Hodgkin lymphoma compared with the general population, respectively. Furthermore, patients with PN-MPN harbouring the JAK2 V617F mutation had a SIR of 5.46 (95% CI: 2.45–12.15) of acquiring a secondary haematolymphoid malignancy [12]. In a Danish population-based cohort study, the SIR of developing a haematologic malignancy in patients with ET was 5 (95% CI: 3.5–6.9) [13]. The SIR in patients with ET was approximately equal to PV in the study and reported as 1.8 (95% CI: 1.1–2.7) and 0.6 (95% CI: 0.2–1.5) in patients with non-Hodgkin lymphoma and lymphoid leukaemia, respectively [13]. While it is well documented that haematolymphoid malignancies may lead to secondary myelofibrosis, PMF with a secondary haematolymphoid malignancy is rare and its occurrence is primarily isolated to case reports.
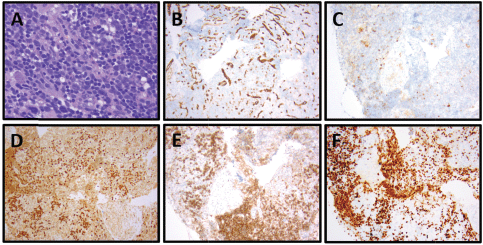
Figure 4. Patient 3, cervical lymph node with T-ALL. (A) Relatively monomorphic infiltrate of neoplastic lymphocytes with irregularly shaped, hyperchromatic, small to medium-sized nuclei with inconspicuous nucleoli, 400x. (B) CD34-negative, (CD34-positive blood vessels as internal control), 100x. (C) CD10-negative, 100x. (D) TdT-positive (nuclear), 100x. (E) CD1a-positive, 100x. (F) Ki-67 with 80% proliferation index, 100x.
The pathophysiology by which a lymphoid transformation occurs in patients with PN-MPN arises has not been clearly established. However, mutations involving JAK2 V617F have been suggested as a potential contributor. There is evidence implicating lympho-myeloid progenitor cells as the cell of origin that culminates in PN-MPN [14]. The JAK-STAT pathway plays a fundamental role in lymphoid proliferation, survival and differentiation [15] and mutations have been observed in paediatric acute lymphoblastic leukaemias, adult T-cell lymphoblastic leukaemias and Hodgkin lymphoma [16, 17]. In addition, there are reported cases of the JAK2 V617F mutation in both PN-MPN as well as lymphoma cells following a lymphomatous transformation, suggestive of a causal link and mutational aetiology for the lymphomatous transformation [12]. Larsen et al [14] discovered a subpopulation of B and T lymphocytes with the JAK2 V617F mutation in a population of patients with PN-MPN, suggesting that this mutation occurs in an early stem cell with both myeloid and lymphoid differentiation potential. This may indicate that there exists a proportion of T-lymphocyte sub-clone precursors arising from a common early lympho-myeloid progenitor that harbour the mutation, creating a predisposition for eventual lymphomatous or leukaemic progression. An assessment of JAK2 mutations was not done in the malignancies in these reported cases and given that the first patient had a triple-negative ET, there may be other mutations harboured by early progenitor cells that were not assessed.
Patient 1 developed AITL 6 months after the diagnosis of triple-negative ET. However, a triple-negative status does not exclude other less common genomic aberrancies [18] which may have also have contributed to the development of a secondary T-cell neoplasm. In 68 patients with previously diagnosed triple-negative ET, Zu et al [19] conducted targeted genomic sequencing and found other mutations in FLT3, SH2B3, ASXL1, ADAMTS1, TET2, TP53, EGFR, CUX1, GATA2 and MPL that may have contributed to the development of ‘triple-negative’ ET. Several of these mutations are also seen in T-cell malignancies. In T-ALL, mutations in FLT3, SH2B3 and ASXL1 have been demonstrated [20, 21], and in AITL, mutations in TET2, TP53 and GATA2 (reported in EBV-associated T-cell lymphoma) have been reported [22–24]. These were not assessed in patient 1, but may have been contributing factors to disease pathogenesis.
Conclusion
This case series demonstrates the possibility of developing T-cell neoplasms in patients with PN-MPN, independent of cytogenetic abnormalities and mutational status. Disease progression may be rapid, and disease recurrence may occur if remission is achieved. Clinicians managing patients with PN-MPN should be aware of this possibility and maintain a high index of suspicion for this potential complication when patients present with ‘B’ symptoms or new LAD. A detailed diagnostic workup should be conducted efficiently so that systemic therapy can be initiated expediently. Workup in T and B-cell malignancies that develop in patients with PN-MPN should include a mutation analysis of JAK2 V617F, CALR and MPL.
Abbreviations
Myeloproliferative neoplasm: MPN; Philadelphia Negative Myeloproliferative Neoplasm: PN-MPN; WHO: World Health Organization; Polycythemia Vera: PV; Essential Thrombocythemia: ET; Primary Myelofibrosis: PMF; Janus Kinase: JAK; Calreticulin: CALR; Thrombopoietin Receptor: MPL.
Conflicts of interest
The authors have no conflicts of interest.
Funding statement
The authors have no financial disclosures to declare.
References
1. Arber DA, Orzai A, and Hasserjian R, et al (2016) The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia Blood 127 2391–2405 https://doi.org/10.1182/blood-2016-03-643544 PMID: 27069254
2. Barbui T, Thiele J, and Gisslinger H, et al (2016) The 2016 revision of WHO classification of myeloproliferative neoplasms: clinical and molecular advances Blood Rev 30(6) 453–459 https://doi.org/10.1016/j.blre.2016.06.001 PMID: 27341755
3. Nangalia J and Green AR (2017) Myeloproliferative neoplasms: from origins to outcomes Blood 130 2475–2483 https://doi.org/10.1182/blood-2017-06-782037 PMID: 29212804
4. Yogarajah M and Tefferi A (2017) Leukemic transformation in myeloproliferative neoplasms. A literature review on risks, characteristics, and outcome Mayo Clin Pro 92(7) 1118–1128 https://doi.org/10.1016/j.mayocp.2017.05.010
5. Barbui T, Thiele J, and Passamonti F, et al (2011). Survival and disease progression in essential thrombocythemia are significantly influenced by accurate morphologic diagnosis: an international study J Clin Oncol 29 3179–3184 https://doi.org/10.1200/JCO.2010.34.5298 PMID: 21747083
6. Tefferi A, Rumi E, and Finazzi G, et al (2013) Survival and prognosis among 1545 patients with contemporary polycythemia vera: an international study Leukemia 27 1874–1881 https://doi.org/10.1038/leu.2013.163 PMID: 23739289 PMCID: 3768558
7. Abdulkarim K, Girodon F, and Johansson P, et al (2009) AML transformation in 56 patients with Ph- MPD in two well defined populations Eur J Haematol 82 106–111 https://doi.org/10.1111/j.1600-0609.2008.01163.x PMID: 19134023
8. Cervantes F, Tassies D, and Salgado C, et al (1991) Acute transformation in nonleukemic chronic myeloproliferative disorders: actuarial probability and main characteristics in a series of 218 patients Acta Haematol 85 124–127 https://doi.org/10.1159/000204873 PMID: 2042444
9. Tam CS, Nussenzveig RM, and Popat U, et al (2008) The natural history and treatment outcome of blast phase BCR-ABL− myeloproliferative neoplasms Blood 112 1628–1637 https://doi.org/10.1182/blood-2008-02-138230 PMID: 18566326 PMCID: 2518875
10. Tefferi A, Guglielmelli P, and Larson DR, et al (2014) Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis Blood 124(16) 2507–2513 quiz 2615 https://doi.org/10.1182/blood-2014-05-579136 PMID: 25037629 PMCID: 4199952
11. Aitchison R, Black AJ, and Greaves MF (1987) Polycythaemia rubra vera transforming to acute lymphoblastic leukaemia Clin Lab Haematol 9 201–204 https://doi.org/10.1111/j.1365-2257.1987.tb01402.x PMID: 3476236
12. Vannucchi AM, Masala G, and Antonioli E, et al (2009) Increased risk of lymphoid neoplasms in patients with Philadelphia chromosome-negative myeloproliferative neoplasms Cancer Epidemiol Biomarkers Prev 18(7) 2068–2073 https://doi.org/10.1158/1055-9965.EPI-09-0353 PMID: 19531676
13. Frederiksen H, Farkas DK, Christiansen CF, et al (2011) Chronic myeloproliferative neoplasms and subsequent cancer risk: a Danish population based cohort study Blood 118(25) 6515–6120 https://doi.org/10.1182/blood-2011-04-348755 PMID: 22039256
14. Larsen TS, Christensen JH, Hasselbalch HC, et al (2007) The JAK2 V617F mutation involves B- and T-lymphocyte lineages in a subgroup of patients with Philadelphia-chromosome negative chronic myeloproliferative disorders Br J Haematol 136(5) 745–751 https://doi.org/10.1111/j.1365-2141.2007.06497.x PMID: 17313377
15. Murray PJ (2007) The JAK-STAT signaling pathway: input and output integration J Immunol 178(5) 2623–2629 https://doi.org/10.4049/jimmunol.178.5.2623 PMID: 17312100
16. Vainchenker W and Constantinescu SN (2013) JAK/STAT signaling in hematological malignancies Oncogene 32(21) 2601–2613 https://doi.org/10.1038/onc.2012.347
17. Roncero AM, López-Nieva P, and Cobos-Fernández MA, et al (2016) Contribution of JAK2 mutations to T-cell lymphoblastic lymphoma development Leukemia 30(1) 94–103 https://doi.org/10.1038/leu.2015.202 PMCID: 4705429
18. Elnahass Y, Mahmoud HK, and Mattar M, et al (2016) Triple negative myeloproliferative neoplasms (MPNs) patients show low MPN10 score and lower grades of bone marrow fibrosis Blood 128 5477 https://doi.org/10.1182/blood.V128.22.5477.5477
19. Ju MK, Fu RF, and Li HY, et al (2018) Clinical characteristic of “triple-negative” essential thrombocythaemia patients and mutation analysis by targeted sequencing Zhongguo Shi Yan Xue Ye Xue Za Zhi 26(4) 1137–1145
20. Zhang J, Ding L, and Holmfeldt L, et al (2012) The genetic basis of early T-cell precursor acute lymphoblastic leukaemia Nature 481(7380) 157–163 https://doi.org/10.1038/nature10725 PMID: 22237106 PMCID: 3267575
21. Prebet T, Carbuccia N, and Raslova H, et al (2013) Concomitant germ-line RUNX1 and acquired ASXL1 mutations in a T-cell acute lymphoblastic leukemia Eur J Haematol 91(3) 277–279 https://doi.org/10.1111/ejh.12147 PMID: 23692290
22. Lemonnier F, Couronné L, and Parrens M, et al (2012) Recurrent TET2 mutations in peripheral T-cell lymphomas correlate with TFH-like features and adverse clinical parameters Blood 120(7) 1466–1469 https://doi.org/10.1182/blood-2012-02-408542 PMID: 22760778
23. Heavican TB, Bouska A, and Yu J, et al (2019) Genetic drivers of oncogenic pathways in molecular subgroups of peripheral T-cell lymphoma Blood https://doi.org/10.1182/blood-2018-09-872549
24. Cohen JI, Dropulic L, and Hsu AP, et al (2016) Association of GATA2 deficiency with severe primary epstein-barr virus (EBV) infection and EBV-associated cancers Clin Infect Dis (1) 41–7 https://doi.org/10.1093/cid/ciw160 PMID: 27169477 PMCID: 4901862





