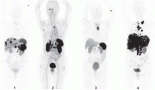Should we perform baseline NGS testing in precursor T lymphoblastic leukaemias: a single centre experience from Eastern India
Prateek Das1,2, Sujeet Kumar2,3, Raghwesh Ranjan2,4, Pradeep Arumugam1,2, Nilesh Dhole2,5, Rohit Kumar Kori2,5, Anil Yadav2,6, Anil Singh2,3, Vikramjit Kanwar2,4 and Neha Singh1,2
1Hematopathology (Oncopathology), Homi Bhabha Cancer Hospital, Varanasi 221010, India
2Homi Bhabha National Institute, Mumbai 400094, India
3Department of Medical Oncology (Adult Hematolymphoid Unit), Homi Bhabha Cancer Hospital, Varanasi 221010, India
4Department of Pediatric Oncology, Homi Bhabha Cancer Hospital, Varanasi 221010, India
5Hematopathology, Homi Bhabha Cancer Hospital, Varanasi 221010, India
6Cancer Cytogenetics, Homi Bhabha Cancer Hospital, Varanasi 221010, India
Abstract
Introduction: T-lymphoblastic leukaemia accounts for approximately one-fourth of acute lymphoblastic leukaemia cases. Sequencing approaches have identified >100 genes that can be mutated in T-cell acute lymphoblastic leukaemia (T-ALL). However, the revised WHO 2022 edition of lymphoid neoplasms still does not incorporate molecular signatures into the T-ALL subgrouping unlike B-ALLs and acute myeloid leukemia, which are classified mainly based on molecular landscapes.
Methods: This retrospective observational study included all newly diagnosed patients of T-lymphoblastic leukaemia of all age groups who presented during the period between January 2022 and October 2023 in whom complete baseline diagnostic work-up was available including flow cytometry, fluorescence in situ hybridization and next generation sequencing studies.
Results: There was a lower frequency of karyotypic abnormalities in adult early T progenitor (ETP)-ALLs than in other sub-groups. Non-ETP ALLs showed significant association with NOTCH1 mutations (p ≤ 0.00001), followed by JAK3 (p = 0.01), FBXW7 (p = 0.066) and PHF6 (p = 0.09) mutations. There was no difference between adult and pediatric patients, in terms of genomic profiling except in the PHF6 gene. There was no significant difference between NOTCH1-mutated and NOTCH1-wild T-ALL patients as well as NOTCH1-heterodimerization versus NOTCH1-PEST mutated patients in terms of measurable residual disease (MRD), relapse-free survival (RFS) and/or overall survival (OS). 45.1% of all TALL patients harboured ≥3 mutations. However, the complex molecular profile did not correlate significantly with MRD positivity and poor RFS and/or OS rates.
Conclusion: Molecular profiling of TALLs do not significantly impact long-term survival outcomes. In resource-constrained settings, we can get away by not doing comprehensive molecular profiling of TALLs at baseline and restrict the sequencing assay to only those cases that are persistently MRD positive or have relapsed.
Keywords: next generation sequencing, minimal residual disease, precursor T- lymphoblastic leukaemia, cytogenetics, mutations, relapse
Correspondence to: Neha Singh
Email: drnehasingh123@gmail.com
Published: 06/12/2024
Received: 05/08/2024
Publication costs for this article were supported by ecancer (UK Charity number 1176307).
Copyright: © the authors; licensee ecancermedicalscience. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Introduction
T-cell acute lymphoblastic leukaemia (T-ALL) is an uncommon, yet aggressive leukaemia that accounts for approximately one-fourth of acute lymphoblastic leukaemia (ALL) cases. Early studies suggested that Early T cell Precursor (ETP)-ALL is a high-risk disease with a high likelihood of induction failure, chemoresistance, high levels of measurable residual disease (MRD), frequent relapses and inferior clinical outcomes [1, 2]. However, subsequent studies using a risk-adapted approach with more intensified therapy demonstrated conflicting results: some showed no significant differences in event-free survival or overall survival (OS) between ETP-ALL and other T-ALLs, whereas others continued to show poor OS [3–8].
Sequencing approaches focusing on candidate oncogenes, or more recently, genome-wide sequencing, have identified >100 genes that can be mutated in T-ALL. Understanding the molecular aberrations may lead to more targeted therapies and improvement of clinical outcomes [9, 10]. However, the revised WHO classification of lymphoid neoplasms still does not incorporate molecular signatures into the T-ALL subgrouping (ETP versus Non-ETP) unlike B-ALLs and acute myeloid leukemia (AML), which are classified mainly based on molecular landscapes [11, 12]. With this in the background, the primary aim of the study was to study the molecular landscape of our cohort of patients with T-lymphoblastic Leukaemia at diagnosis and their correlation with T-MRD studies and survival outcomes.
Materials and methods
This retrospective observational study included all newly diagnosed patients of T-lymphoblastic leukaemia of all age groups who presented during the period between January 2022 and October 2023 in whom complete baseline diagnostic work-up was available including flow cytometry, fluorescence in situ hybridization (FISH) and next generation sequencing (NGS) studies. Patients with partially-treated TALL from outside as well as relapsed TALLs were excluded from this study. Patients were categorised in accordance with the revised 2022 WHO classification of lymphoid neoplasms. Ethical clearance was obtained for the conduct of the study.
FISH studies were performed on heparinised bone marrow aspirate or peripheral blood of patients using commercially available disease-specific probes (deletion, break-apart and centromeric) according to the manufacturer’s protocol.
For NGS assay, DNA extracted from bone marrow/peripheral blood sample was used as a template. DNA was fragmented which was followed by end repair, A tailing and adapter ligation. Whole genome libraries with patient-specific indices were amplified, followed by hybridization with 1,313 hybrid capture oligonucleotide probes to sequence a 247.74 kb panel of 70 genes. The genes include ABL1 (exons 1–11), ANKRD26 (exons 1–34), ASXL1 (exons 1–13), ASXL2 (exons 1–11), ATM (exons 2–63), ATRX (exons 1–35), BCOR (exons 2–15), BCORL1(exons 2–15), BIRC3 (exons 2–9), BRAF (exons 1–18), BRINP3 (exons 2–7), CALR (exons 1–9), CBL (exons 1–16), CBLB (exons 1–19), CBLC (exons 1–10), CDKN2A (exons 1–3), CEBPA (exon 1), CSF3R (exons 3–18), CUX1 (exons 1–21), DDX41 (exons 1–16), DNMT3A (exons 1–18), ETNK1 (exons 1–8), ETV6 (exons 1–8), EZH2 (exons 2–20), FLT3 (exons 1–24), FBXW7 (exons 1–11), GATA1 (exons 2–6), GATA2 (exons 2–7), GNAS (exons 1–13), HRAS (exons 2–5), IDH1 (exons 1–10), IDH2 (exons 1–9), IKZF1 (exons 2–7), JAK2 (exons 2–25), JAK3 (exons 2–24), KDM6A (exons 1–30), KIT (exons 1–21), KMT2A (exons 2–36), KRAS (exons 2–5), MPL (exons 1–12), MYD88 (exons 1–5), NF1 (exons 1–57), NOTCH1 (exons 1–34), NPM1 (exons 1–12), NRAS (exons 2–5), PDGFRA (exons 2–24), PHF6 (exons 2–10), PIGA (exons 1–6), POT1 (exons 5–18), PPM1D (exons 1–6), PTEN (exons 1–9), PTPN11 (exons 1–15), RAD21 (exons 2–14), RET (exons 1–15,19,20), RUNX1 (exons 2–6), SETBP1 (exons 2–6), SF3B1 (exons 1–20), SH2B3 (exons 1–15), SMC1A (exons 1–26), SMC3 (exons 1–29), SRSF2 (exons 1–2), STAG1 (exons 2–34), STAG2 (exons 2–35), TET2 (exons 3–11), TP53 (exons 1–11), U2AF1 (exons 1–8), WT1 (exons 1–9), XPO1 (exons 2–25), ZBTB7A (exons 2–3) and ZRSR2 (exons 1–11). Sequencing was performed on Illumina MiniSeq using the P2 150-cycle chemistry. Bioinformatics was obtained by Illumina-based solutions. Finally, the variants were annotated and prioritised with population frequency databases (1,000 G (African, Admixed American, East Asian, Finnish, Non-finnish European, South Asian), Exome Aggregation Consortium datasets, NHLBI-ESP (6,500 genomes)) and effect-based databases including ClinVar, Ensembl VEP, Franklin and Varsome as well as the COSMIC databases.
Diagnostic and post-induction MRD immunophenotyping was performed on bone marrow aspirate sample using bulk lysis staining method and further acquired on 3-laser, 13-color Dx FLEX flowcytometer (Beckman Coulter, USA) using cyt expert acquisition software. The 10-color antibody panel for T-MRD studies is shown in Supplementary Table 1. At least 100,000 events were acquired during diagnostic work-up and 1 million events during MRD assessment in each tube. Note that only the first pull bone marrow aspirate was submitted for Measurable residual disease detected by multi-parametric flowcytometry. Immunophenotyping data was analysed with Kaluza (v 2.1) software (Beckmann Coulter, USA), using a pre-defined template-based approach (Supplementary Figure 1).
Patients were treated with Intermediate-risk Berlin Frankfurt Munich (BFM)-95 protocol in adults ≥15 years and High-risk-ICiCLe protocol in pediatric patients <15 years. MRD assessment was done by flow cytometry post-induction (after Induction IA in BFM-95) and post-consolidation (after Induction IIA in BFM-95 protocol). Pediatric patients went for post-consolidation MRD studies only if PI-MRD was positive. The adult patients who were MRD positive after consolidation were counselled for high-risk BFM-95 protocol and stem cell transplant. However, none of the patients opted for bone marrow transplantation due to financial constraints or non-availability of a suitable donor. They were hence continued on BFM protocol, if morphological bone marrow remission was achieved (BM blasts <5% confirmed by flow cytometry), or else they were treated with palliative intent and taken off BFM protocol. Relapse-free survival (RFS) was calculated from the date of complete remission (CR) until the time to relapse or death or last follow-up if in CR.
Table 1. Baseline demographic and hematologic characteristics of T-ALL patients at presentation.
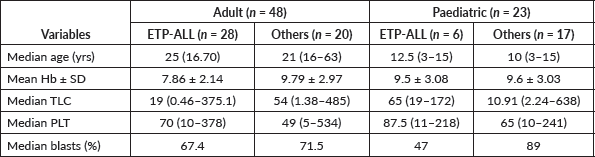
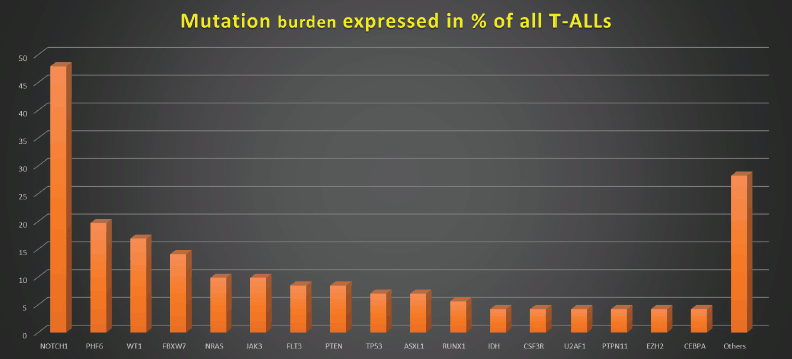
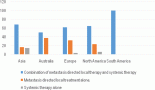
Figure 1. Mutation burden expressed in % of all T-ALLs.
Data were analysed by using SPSS Software version 23 and presented in median (minimum–maximum), and frequency percentage. The association between categorical variables was tested by the Fisher exact test and chi-square test. All tests were 2-sided and p value <0.05 was considered statistically significant. Univariate analysis of the MRD assays and cytogenetic and molecular risk groups was analysed for their impact on RFS using the Kaplan–Meier technique and compared using the log-rank test.
Results
Out of a total of 71 newly diagnosed T-ALLs fulfilling the inclusion criteria, 48 were adults and 23 were pediatric patients. The frequency of ETP-ALLs was higher among adults (58.3% versus 26.1%) and was associated with severe anemia and neutropenia. Their detailed demographic and baseline hematologic features have been shown in Table 1.
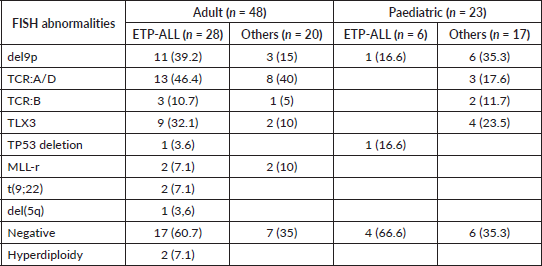
Among the adult T-ALLs, T cell receptor (TCR): A/D rearrangement was the commonest abnormality detected by FISH, unlike the pediatric cohort, in which del9p followed by TLX3 rearrangements were more evident. There was a lower frequency of karyotypic abnormalities in adult ETP-ALLs than in other sub-groups (Supplementary Table 2). As shown in Table 2, ETP-ALLs showed a trend towards higher chances of positive PI-MRD >0.01% (p = 0.066) in adult patients and steroid refractoriness (Day +8 blast count) in pediatric patients (p = 0.056); however, there was no significant difference between different age groups and WHO-defined subgroups in terms of induction failure rates, RFS or OS.
Figures 1–3 show the diagrammatic representation of the mutational spectrum and frequency of all TALL patients as well as in ETP-ALLs versus Other TALL subgroups, which were not found to be significantly different across all age groups except NOTCH1 mutations as shown in Table 2. Overall, other ALLs showed significant association with NOTCH1 mutations (p ≤ 0.00001), followed by JAK3 (p = 0.01), FBXW7 (p = 0.066) and PHF6 (p = 0.09) mutations. On the contrary, ETP-ALLs showed a predilection towards molecular signatures involved in AML leukemogenesis such as FLT3, NRAS, WT1 and TP53. There was no difference between adult and pediatric patients also, in terms of genomic profiling except in PHF6 gene. PHF6 mutations commonly involved exon 8 followed by exons 2, 7 and 9 and were frame-shift insertions or stop-gain mutations in adult patients, unlike the high frequency of mis-sense mutations involving exon 9 in pediatric patients. The VAF% varied between 8.3% and 90.9%.
NOTCH1 mutations occurred at an overall frequency of 47.9% (34/71) in our TALL patients, irrespective of age or subtypes. NOTCH1 gene mutations mainly affected the heterodimerization (HD) domain involving exons 26 and 27 (58.8%; 20/34) and PEST domain involving exon 34 (29.4%; 10/34). 11.8% NOTCH1-mutated patients involved both HD and PEST domains. Mutations were mostly frameshift insertions and/or mis-sense point substitutions. VAF% in NOTCH-mutated patients varied from 5.83% to 51.9%. Two patients showed multiple NOTCH1 mutations. Two patients showed induction failure in each of the three NOTCH-mutated subgroups, i.e., HD, PEST and both. Four patients with NOTCH1-HD mutations and one patient with NOTCH1-PEST mutation had >0.01% MRD-PI positivity, respectively. Four NOTCH1-mutated patients were MRD positive while ten NOTCH1-unmutated patients were MRD positive (p value = 0.1). None of the NOTCH mutated patients showed relapse or died. There was no significant difference between NOTCH1-mutated and NOTCH1-wild T-ALL patients as well as NOTCH1-HD versus NOTCH1-PEST mutated patients in terms of MRD, RFS and/or OS.
Table 2. Prognostic factors in T-ALL.
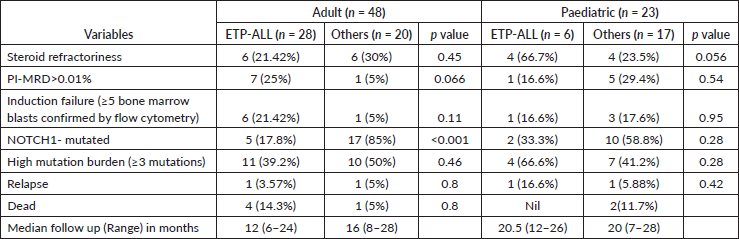
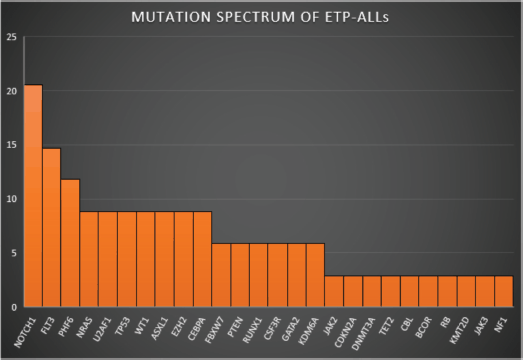
Figure 2. Mutation spectrum of ETP-ALLs.
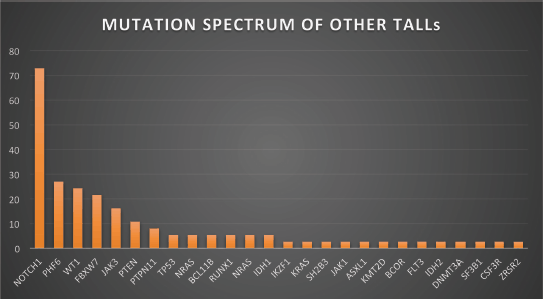
Figure 3. Mutation spectrum of other TALLs.
The molecular spectrum of TALLs is complex which can be elucidated by the fact that 45.1% of all patients harboured ≥3 mutations; however, on univariate analysis, the complex molecular profile did not correlate significantly with MRD positivity and poor RFS and/or OS rates.
Discussion
We retrospectively analysed the molecular landscape of TALL patients and tried to find out the correlation with MRD and/or treatment outcomes. Our ETP-ALL patients were enriched in myeloid-associated mutations such as FLT3, NRAS, WT1, U2AF1 and TP53, with relatively reduced frequency of gene mutations typically seen in other subtypes of T-ALL, such as NOTCH1, FBXW7 and JAK3, similar to the findings of Ye et al [13]. We found no association between MRD positivity, RFS and/or OS and the mutation status of genes assessed including NOTCH1, PHF6, FLT3 and WT1 and also detected no differences in survival between patients with one or two mutations and patients with three or more mutations similar to the results of their study. The only difference was in the higher mutation burden in their ETP ALL patients and adverse prognostic impact of TP53 mutation in comparison to our study in which both ETP and Non-ETP patients had equivalent mutational burden (Table 2), and prognostic significance of TP53 mutation could not be ascertained due to paucity of TP53 mutations [13, 14].
85.9% of our patients had at least one mutation, and 45.1% had ≥3 mutations. The genes with higher mutation frequency were NOTCH1, PHF6 and WT1, in comparison to the results of a study by Yin et al [15] in which 73.3% of patients had at least one mutation, and 36.7% had ≥3 mutations. The genes with higher mutation frequency in their study were NOTCH1, FBXW7 and DNMT3A [15].
NOTCH1 gene consists of extracellular N-terminal epidermal growth factor-like repeats, LIN-12/Notch related repeat domains, HD and C-terminal intracellular domains with PEST motif. Some authors reported that NOTCH1 mutations correlated with favourable long-term outcomes in paediatric T-ALL [16, 17] while others found no association of NOTCH1 mutation status with treatment outcomes [13]. We did not observe a correlation between NOTCH1 mutations and survival outcomes. Our findings were in striking contrast to that of Yuan et al [18] and Burns et al [19] who identified NOTCH1 gene status and MRD post-induction as independent prognostic factors and favoured inclusion of NOTCH1 mutation status as a risk stratification factor in TALL. The discrepancy may be due to different types of mutations, different ethnic populations and different treatment regimens.
NOTCH1 mutations in our study were located at either HD domain involving exons 26 and 27 (58.8%) or PEST domain involving exon 34 (29.4%). 11.8% NOTCH1-mutated patients involved both HD and PEST domains. The only Indian study concerning the same agenda by Bhatia et al [20] showed NOTCH1 mutations in 52% of their pediatric TALLs, of which 71% were in HD domain and 35% in PEST domain (including one case with mutations in all three domains). They showed that cases with PEST domain NOTCH mutations had poor RFS but the OS was not influenced by NOTCH mutation positivity. We differ on this aspect as RFS and/or OS was not affected by HD or PEST domain of NOTCH mutations in our patients.
PHF6 gene is an X-linked tumour suppressor involved in the pathogenesis of T-ALL. In T-ALL, there have been mixed reports regarding its prognostic roles; some demonstrated inferior outcomes in patients with mutated PHF6 [21] whereas others showed no significant differences in outcomes from those with wild-type PHF6 [22, 23]. We detected PHF6 mutations in 11.8% of our ETP-ALLs and 27% of Other TALL cases, including nonsense and frameshift mutations in adults and mis-sense mutations in pediatric patients and all occurred in conjunction with other mutations, such as NOTCH1 and WT1. No impact on OS was observed, again likely compounded by the relatively small sample size in the current study [13].
The WT1 gene is mutated in approximately 10% of TALL cases, and comprised of mainly heterozygous frameshift mutations that cluster in exon 7 and are predicted to lead to a truncated protein Moreover, WT1 is shown to be among the frequently altered genes in the ETP ALL subgroup. In T-ALL, studies on the role of WT1 mutations are still limited. In pediatric and adult T-ALL, the presence of WT1 mutations have not been predictive of poor clinical outcome by Bordin et al [24]. In our cohort, WT1 mutations were clustered in exons 7, 8 and 13 and were mostly frameshift indels. The overall frequency of WT1 mutations in TALLs was higher (16.9%) in comparison to their study. Also, the frequency in other TALLs (24.3%) was higher than in ETP-ALLs (8.8%) in our cohort contrary to their findings. However, we also could not establish any impact of WT1 mutations in TALLs on survival outcomes irrespective of age or subtype.
On the basis of the above findings, it may be right to say that molecular profiling of TALLs do not significantly impact long-term survival outcomes, similar to most recently published literature and hence does not find a place in the WHO revised classification of TALLs. That may also be due to the small sample size and short follow-up duration which is the only limitation of this study. In resource-constrained settings, we can get away by not doing comprehensive molecular profiling of TALLs at baseline and restrict the sequencing assay to only those cases that are persistently MRD positive or have relapsed. Also, the intensive treatment regimens nullify the effects of complex molecular landscape to a great extent. It is the largest study on TALL molecular landscape from India and more such studies will pave the way for a clear understanding of TALL biology of Asian patients.
Conflicts of interest
The authors state that there is no conflict of interest present.
Funding
NA.
Consent for publication
It is confirmed that this work is original and has not been published elsewhere, nor is it currently under consideration for publication elsewhere. The manuscript has been read and approved by all the authors.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
References
1. Coustan‐Smith E, Mullighan CG, and Onciu M, et al (2009) Early T‐cell precursor leukaemia: a subtype of very high‐risk acute lymphoblastic leukaemia Lancet Oncol 10(2) 147–156 https://doi.org/10.1016/S1470-2045(08)70314-0
2. Neumann M, Heesch S, and Gökbuget N, et al (2012) Clinical and molecular characterization of early T‐cell precursor leukemia: a high‐risk subgroup in adult T‐ALL with a high frequency of FLT3 mutations Blood Cancer J 2(1) e55 https://doi.org/10.1038/bcj.2011.49 PMID: 22829239 PMCID: 3270253
3. Patrick K, Wade R, and Goulden N, et al (2014) Outcome for children and young people with early T‐cell precursor acute lymphoblastic leukaemia treated on a contemporary protocol, UKALL 2003 Br J Haematol 166(3) 421–424 https://doi.org/10.1111/bjh.12882 PMID: 24708207
4. Bond J, Graux C, and Lhermitte L, et al (2017) Early response‐based therapy stratification improves survival in adult early thymic precursor acute lymphoblastic leukemia: a group for research on adult acute lymphoblastic leukemia study J Clin Oncol 35(23) 2683–2691 https://doi.org/10.1200/JCO.2016.71.8585 PMID: 28605290
5. Jain N, Lamb AV, and O’Brien S, et al (2016) Early T‐cell precursor acute lymphoblastic leukemia/lymphoma (ETP‐ALL/LBL) in adolescents and adults: a high‐risk subtype Blood 127(15) 1863–1869 https://doi.org/10.1182/blood-2015-08-661702 PMID: 26747249 PMCID: 4915808
6. Morita K, Jain N, and Kantarjian H, et al (2021) Outcome of T‐cell acute lymphoblastic leukemia/lymphoma: focus on near‐ETP phenotype and differential impact of nelarabine Am J Hematol 96(5) 589–598 https://doi.org/10.1002/ajh.26144 PMID: 33639000
7. Noronha EP, Marques LVC, and Andrade FG, et al (2019) The profile of immunophenotype and genotype aberrations in subsets of pediatric T-cell acute lymphoblastic leukemia Front Oncol 9 316 https://doi.org/10.3389/fonc.2019.00316 PMID: 31338319 PMCID: 6503680
8. Schrappe M, Valsecchi MG, and Bartram CR, et al (2011) Late MRD response determines relapse risk overall and in subsets of childhood T-cell ALL: results of the AIEOP-BFM-ALL 2000 study Blood 118 2077–2084 https://doi.org/10.1182/blood-2011-03-338707 PMID: 21719599
9. Li Z, Song Y, and Zhang Y, et al (2020) Genomic and outcome analysis of adult T-cell lymphoblastic lymphoma Haematologica 105(3) e107–e110 https://doi.org/10.3324/haematol.2019.220863 PMCID: 7049353
10. Girardi T, Vicente C, and Cools J, et al (2017) The genetics and molecular biology of T-ALL Blood 129(9) 1113–1123 https://doi.org/10.1182/blood-2016-10-706465 PMID: 28115373 PMCID: 5363819
11. Alaggio R, Amador C, and Anagnostopoulos I, et al (2022) The 5th edition of the World Health Organization classification of haematolymphoid tumours: lymphoid neoplasms Leukemia 36 1720–1748 https://doi.org/10.1038/s41375-022-01620-2 PMID: 35732829 PMCID: 9214472
12. Khoury JD, Solary E, and Abla O, et al (2022) The 5th edition of the World Health Organization classification of haematolymphoid tumours: myeloid and histiocytic/dendritic neoplasms Leukemia 36 1703–1719 https://doi.org/10.1038/s41375-022-01613-1 PMID: 35732831 PMCID: 9252913
13. Ye MT, Wang Y, and Zuo Z, et al (2023) Integrated clinical genotype‐phenotype characteristics of early T‐cell precursor acute lymphoblastic leukemia Cancer 129(1) 49–59 https://doi.org/10.1002/cncr.34515
14. González-Gil C, Morgades M, and Lopes T, et al (2023) Genomics improves risk stratification of adults with T-cell acute lymphoblastic leukemia enrolled in measurable residual disease-oriented trials Haematologica 108(4) 969–980 https://doi.org/10.3324/haematol.2022.281196 PMCID: 10071117
15. Yin H, Hong M, and Deng J, et al (2022) Prognostic significance of comprehensive gene mutations and clinical characteristics in adult T-cell acute lymphoblastic leukemia based on next-generation sequencing Front Oncol 12 811151 https://doi.org/10.3389/fonc.2022.811151 PMID: 35280829 PMCID: 8908046
16. Kox C, Zimmermann M, and Stanulla M, et al (2010) The favorable effect of activating NOTCH1 receptor mutations on long‐term outcome in T‐ALL patients treated on the ALL‐BFM 2000 protocol can be separated from FBXW7 loss of function Leukemia 24(12) 2005–2013 [doi:10.1038/leu.2010.203] https://doi.org/10.1038/leu.2010.203 PMID: 20944675 PMCID: 3035973
17. Breit S, Stanulla M, and Flohr T, et al (2006) Activating NOTCH1 mutations predict favorable early treatment response and long‐term outcome in childhood precursor T‐cell lymphoblastic leukemia Blood 108(4) 1151–1157 [doi:10.1182/blood‐2005‐12‐4956] https://doi.org/10.1182/blood-2005-12-4956 PMID: 16614245
18. Yuan Y, Li J, and Xue TL, et al (2022) Prognostic significance of NOTCH1/FBXW7 mutations in pediatric T cell acute lymphoblastic leukemia: a study of minimal residual disease risk-directed CCLG-ALL 2008 treatment protocol Leuk Lymphoma 63(7) 1624–1633 https://doi.org/10.1080/10428194.2022.2032033 PMID: 35129045
19. Burns MA, Place AE, and Stevenson KE, et al (2021) Identification of prognostic factors in childhood T-cell acute lymphoblastic leukemia: results from DFCI ALL consortium protocols 05-001 and 11-001 Pediatr Blood Cancer 68(1) e28719 https://doi.org/10.1002/pbc.28719
20. Bhatia P, Totadri S, and Singh M, et al (2020) PEST domain NOTCH mutations confer a poor relapse free survival in pediatric T-ALL: Data from a tertiary care centre in India Blood Cells Mol Dis 82 102419 https://doi.org/10.1016/j.bcmd.2020.102419 PMID: 32179411
21. Yeh TC, Liang DC, and Liu HC, et al (2019) Clinical and biological relevance of genetic alterations in pediatric T‐cell acute lymphoblastic leukemia in Taiwan Pediatr Blood Cancer 66(1) e27496 https://doi.org/10.1002/pbc.27496
22. Van Vlierberghe P, Palomero T, and Khiabanian H, et al (2010) PHF6 mutations in T‐cell acute lymphoblastic leukemia Nat Genet 42(4) 338–342 https://doi.org/10.1038/ng.542 PMID: 20228800 PMCID: 2847364
23. Wang Q, Qiu H, and Jiang H, et al (2011) Mutations of PHF6 are associated with mutations of NOTCH1, JAK1 and rearrangement of SET‐ NUP214 in T‐cell acute lymphoblastic leukemia Haematologica 96(12) 1808–1814 https://doi.org/10.3324/haematol.2011.043083 PMID: 21880637 PMCID: 3232263
24. Bordin F, Piovan E, and Masiero E, et al (2018) WT1 loss attenuates the TP53-induced DNA damage response in T-cell acute lymphoblastic leukemia Haematologica 103(2) 266–277 https://doi.org/10.3324/haematol.2017.170431 PMCID: 5792271
Supplementary tables and figure
Supplementary Table 1. 10- color antibody panels used for bone marrow TALL-MRD assessment.

Supplementary Table 2. FISH abnormalities in different T-ALL categories.
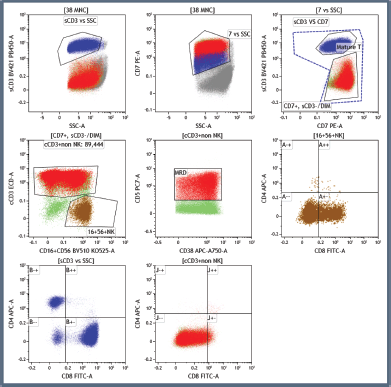
Supplementary Figure 1. Illustration of the T-MRD positive population marked in red, differentiating them from NK cells, mature T cells and myeloblasts in a non-ETP-ALL patient.