Oncolytic virotherapy: new weapon for breast cancer treatment
Veronica Martini1,2a, Francesca D’Avanzo1, Paola Maria Maggiora1, Feba Maria Varughese1,2, Antonio Sica3,4b and Alessandra Gennari1,2c
1Division of Oncology, Department of Translational Medicine, University of Eastern Piedmont, Novara 13100, Italy
2Center for Translational Research on Autoimmune & Allergic Diseases – CAAD, Novara 28100, Italy
3Department of Pharmaceutical Sciences, University of Eastern Piedmont, A Avogadro 28100, Italy
4Department of Inflammation and Immunology, Humanitas Clinical and Research Center–IRCCS, Rozzano (MI) 20089, Italy
ahttps://orcid.org/0000-0002-0887-4082
bhttps://orcid.org/0000-0002-8342-7442
chttps://orcid.org/0000-0002-0928-2281
Abstract
The recent introduction of viruses as a weapon against cancer can be regarded as one of the most intriguing approaches in the context of precision medicine. The role of immune checkpoint inhibitors has been extensively studied in early and advanced cancer stages, with extraordinary results. Although there is a good tolerability profile, especially when compared with conventional chemotherapy, severe immune-related adverse events have emerged as a potential limitation. Moreover, there are still treatment-resistant cases and thus further treatment options need to be implemented. Several in vitro and in vivo studies have been conducted and are ongoing to develop oncolytic viruses (OVs) as a tool to modulate the immune system response. OVs are attenuated viruses that can kill cancer cells after having infected them, producing microenvironment remodelling and antitumour immune response. The potential of oncolytic virotherapy is to contrast the absence of T cell infiltrates, converting ‘cold’ tumours into ‘hot’ ones, thus improving the performance of the immune system. Breast cancer, the second most common cause of cancer-related deaths among women, is considered a ‘cold’ tumour. In this context, oncolytic virotherapy might well be considered as a promising strategy. This review summarises the current status, clinical applications and future development of OVs, focusing on breast cancer treatment.
Keywords: OVs, immunotherapy, BC
Correspondence to: Alessandra Gennari
Email: alessandra.gennari@uniupo.it
Published: 03/12/2020
Received: 11/06/2020
Publication costs for this article were supported by ecancer (UK Charity number 1176307).
Copyright: © the authors; licensee ecancermedicalscience. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Background
The immune system plays an important role in controlling and eradicating cancer. However, malignant cells can develop multiple mechanisms of immune suppression [1]. In recent years, a number of immunomodulating agents have been developed as anticancer therapies and introduced into the clinical practice. Among these, immune checkpoint inhibitors (ICIs) have demonstrated an impressive level of activity as antitumour treatment in different tumour types [2], such as advanced melanoma, non-small cell lung cancer and breast cancers [2, 3]. The major mechanism of action involves the removal of inhibitory pathways that block effective anti-tumour T cell responses; as a consequence, the immune system is re-educated to fight against cancer cells. Yet, the activity of this class of agents is heterogeneous within the same tumour type, and innovative and complementary strategies need to be developed. In particular, tumour microenvironment (TME) might exert an important role in conditioning the immune response. In fact, it has been shown that the so-called hot tumours, characterised by the presence of Tumour-Infiltrating Lymphocytes (TILs), expression of programmed death-ligand 1 (PD-L1) on tumour-associated immune cells and possible genomic instability have the higher chance of response [4]. Conversely, ‘cold’ tumours scarcely expressing PD-L1, characterised by low mutational burden (low expression of neoantigens) and low expression of antigen presentation markers, such as major histocompatibility complex class I (MHC I) [4] are considered less responsive. In this scenario, the treatment of immune ‘cold’ tumours, such as tumours of the breast, prostate, ovary, pancreas and others, with this class of agents may be characterised by suboptimal responses and resistance. In this perspective, different approaches have been identified to overcome the absence of T cell infiltrates, thus converting ‘cold’ tumours into ‘hot’ ones. Among these, chemotherapy has been evaluated as a possible strategy to reshape tumour milieu and get immune infiltrate compatible with an expected activity of the ICI treatment [5, 6]. Many studies are also exploring the ability of viruses to kill tumour cells by initiating systemic immune responses through different mechanisms such as induction of Immunogenic Cell Death (ICD), release of danger signals (damage-associated molecular pattern (DAMPs)) and of tumour antigens from virus-infected cells [7].
Oncolytic Viruses (OVs) are native or genetically modified viruses that selectively infect and replicate within tumour cells, eventually leading to tumour cell lysis. Alongside this direct and local antitumour activity, OVs can also induce a potent, systemic and potentially durable antitumour immunity. The dying tumour cells release a lot of factors that engage antitumour immunity, inducing therapeutic responses also at distant tumour sites [7].
Several OVs have been successfully tested in different preclinical models [8]. In 2015, the Food and Drug Administration (FDA) and European Medicines Agency (EMA) approved the use of talimogene laherparepvec (T-VEC, Imlygic®) as oncolytic viral therapy for advanced melanoma patients [9]. In glioma, several OVs have clearly demonstrated both safety and a promising efficacy in the phase I clinical trials [7]. Various OVs, such as HF10 (Canerpaturev—C-REV) and CVA21 (CAVATAK), are now actively being developed in phase II as monotherapies, or in combination with ICI against melanoma [10]. The clinical trials of OVs against pancreatic cancer have not yet demonstrated efficacy as either monotherapy or as part of combination therapy [7].
This review provides an overview on the development of oncolytic virus immunotherapy. In the first part, we briefly describe the mechanisms of immunosurveillance that are the basis on which the OVs are built and developed. In the second part, we discuss preclinical and clinical data that show a role for OVs in breast cancer therapy.
Cancer cells and host immune responses: mechanism of immunosurveillance
TME is composed of many cell types, including the original cancer cell with genetic alterations and other cells, such as fibroblasts, endothelial cells, and eventually a variety of immune cells that can mount a response against tumour cells. Those tumour cells that escape from immune system attack can establish an immunosuppressive microenvironment. This highly dynamic immunoediting process is defined by three phases: (1) the elimination phase (i.e., immunosurveillance), where the malignant cells are detected and cleared by immune cells; (2) the equilibrium/editing phase, where there is a continuous T-cell-mediated eradication of malignant cells via effector responses including CD8 T-cells, γδ T-cell subsets and Natural Killer (NK) cells, as well as macromolecules including interferon g (IFNγ), perforin, and TNF-related apoptosis-inducing ligands and (3) the escape phase, characterised by uncontrolled cell growth and proliferation that can lead to metastatic spread [11]. TILs, tumour-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs) play an important role in the promotion and maintenance of tumour immune escape (Figure 1).
TILs were defined as lymphocytes that moved from the blood into the tumour bed and, directly opposing and/or surrounding tumour cells, can recognise and kill them. They include T, B and NK cells [12]. The interactions between TILs and cancer cells play a critical role in disease progression. Therefore, the role of TILs has been investigated in the TME. CD8 and CD4 T lymphocytes can be activated directly or indirectly by cancer cells: in the first case, tumour cells can present neoantigens by means of MHC-I and MHC-II molecules; alternatively, T cell activation can occur indirectly through dendritic cells (DC) or macrophages (M), killing, processing and presenting neoantigens to T lymphocytes [12]. TILs exert a good prognostic effect in patients treated with ICI (i.e., anti-PD-1/PD-L1 pathway) not only because they activate the antitumour immune response but also because they release IFNg, favouring PD-L1 expression in tumour cells and providing a positive feedback [13]. Activated cytotoxic CD8 T cells and NK cells have been associated with a good prognosis; in contrast, the presence of Treg has a negative effect on several cancers, due to the enhancement of tumour aggressiveness and suppression of antitumour immune responses [14]. In normal conditions, Treg cells are involved in maintaining immune homeostasis by protecting hosts against the development of allergic and autoimmune disorders [15]. In this regard, it has been demonstrated that enhanced presence of Treg cells in biopsy samples of the breast tumours is associated with an adverse relapse-free and overall survival [16]. Breast cancer is not considered to be immunogenic, due to several factors, including a low tumour mutational load [17]. Indeed, specific TIL subtypes have been associated with different outcomes in Triple-Negative Breast Cancer (TNBC) patients [17]: in particular, high levels of PD-1 TILs or FOXP3 TILs have been shown to correlate with a poor prognosis, whereas high levels of CD8 TILs seem to be associated with an improved prognosis [18].
The reduction of effector functions of TILs, the promotion of T-reg and the secretion of inhibitory cytokines are the consequence of TAMs activity, thus promoting tumour growth and progression [19]. TAMs derive from peripheral blood monocytes recruited into the microenvironment and in response to several stimuli, such as inflammation, undergo M1 (classical) or M2 (alternative) activation. It is well known that some cytotoxic agents are able to favour the antitumour activities of TAMs, at least in leukaemia and/or immunogenic (transplant) tumour models [20]. In this regard, chemotherapy can induce ICD which is triggered by innate immune cells or infiltrating immune cells, including TAM and tumour-associated dendritic cells (TADCs). ICD activates TADC and TAM sustaining enhanced antigen presentation and supporting specific anti-tumour immunity [21]. In this scenario, phagocytosis of cancer cells by TAMs provides additional antitumour activity, which, however, is impaired by the ‘don’t eat’ me signal CD47, expressed by many cancer cells [20]. CD47 is a transmembrane glycoprotein with an inhibitory effect on phagocytosis, called ‘don’t eat me’ signal. This is mediated by CD47 binding to signal-regulatory protein (SIRPα), which is expressed on TAMs [22]. CD47 is overexpressed in many types of human cancer cells and protects them from being recognised and cleared by innate immune surveillance. Since high CD47mRNA expression is associated with poor survival of cancer patients [23], antibodies anti-CD47 were studied in various preclinical models and are currently being tested in ongoing clinical trials [24]. Our unpublished preliminary data, obtained by Immunohistochemistry (IHC), showed a major expression of both CD47 and SIRP1a in TNBC with respect to Luminal BC. Moreover, it was demonstrated that treatment with anti-human CD47 of cancer stem cells isolated from the Triple-Negative MDA-MB-231 cell line decreases proliferation and up-regulates tumour suppressor genes [25]. In this scenario, several TAM-centred strategies have been proposed, such as the suppression of TAM recruitment, the depletion of their number, the promotion of M1-polarised TAMs and the inhibition of TAM-associated molecules [26, 27].
Along with TAMs, MDSCs are characterised by the capacity to suppress T cell functions and support tumour progression [28]. MDSCs are a heterogenous group of immune cells from the myeloid lineage, particularly enriched in the peripheral blood, bone marrow (BM), and neoplastic tissues in cancer patients, exerting a suppressive effect on antitumour immunity, by inhibiting both innate and adaptive immune reactions [29]. These cells comprise at least two subsets: monocytic MDSCs (identified as CD11b Ly6G−Ly6Chi cells in mouse and CD11b CD14 HLA−DRlow/−CD15− cells in human) and granulocytic MDSCs (PMN-MDSCs, identified as CD11b Ly6G Ly6Clo cells in mouse and CD11b CD14−CD15 or CD11b CD14−CD66 cells in human) [28]. Accumulation of myeloid progenitors and their differentiation to TAMs and MDSCs is the result of a process driven by cancer-related inflammation, involving: altered myelopoiesis; mobilisation of myeloid precursors from the BM; recruitment of MDSCs and TAMs precursors into both secondary lymphoid organs and/or tumour tissues; functional diversion of myeloid cells in response to microenvironmental signals. This multistep process drives the reprogramming of myeloid cells towards a tumour-promoting phenotype and remotely controls the composition of the tumour-microenvironment. Previous studies demonstrated that a decrease in immunosuppressive MDSCs leads to the activation of host immunity, and that several chemotherapy drugs, including fluorouracil and gemcitabine, reduced the numbers of MDSCs in tumour-bearing hosts [30]. In addition, immunostimulatory molecules, including CpG oligodeoxynucleotide and polyinosinic-polycytidylic acid (polyI:C), which are ligands of toll-like receptors (TLRs) and/or retinoic acid–inducible gene-I (RIG-I)–like receptors, inhibit the immunosuppressive functions of MDSCs and/or induce differentiation of MDSCs into immune-activating cells, including macrophages [31]. MDSC’s function has been studied in breast cancer. These studies demonstrated that MDSCs induce inhibition of T cells, NK cells and DCs, while stimulating immune regulators such as Th2 T cells, T-reg, and TAMs. MDSCs also secrete cytokines such as IL-6 and IL-4 allowing MDSC expansion and subsequent sequestration of essential amino acids such as arginine and cysteine, which are required for the survival of T cells. MDSCs are also thought to mediate their inhibitory functions through reactive oxygen species such as nitric oxide [29].
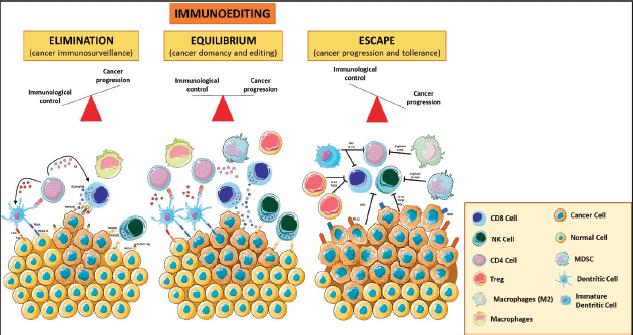
Figure 1. Immunoediting. The three phases of the cancer immunoediting process: elimination, equilibrium and escape. In tumour elimination (left), which often occurs in early tumour development, highly antigenic tumour clones are recognised and eliminated by both innate and adaptive immune systems. In the equilibrium phase (middle), the tumour and the adaptive immune system coexist. Tumour escape (right) occurs when there is poor antigenic expression, immunosuppressive cytokines, MDSCs, and expression of negative regulatory receptors. Various forms of immunotherapy aim to shift the balance from escape and equilibrium to elimination. Treg: T regulatory; MDSCs: Myeloid Derived Suppressor Cells; MCH I: Major Histocompatibility Complex Class I. The figure was modified from Servier Medical Art, http://smart.servier.com/.
Recently, the involvement of extracellular vesicles (EVs) in cancer pathophysiology has been described and, given their release from all cell types under specific stimuli, have also been proposed as potential biomarkers in cancer. In particular, the role of exosomal-PD-L1 fraction has been recently hypothesiSed as a possible determinant of response to ICI, in patients with lung cancer and melanoma [32]. Exosomes are nano-sized membrane vesicles produced in the endosomal cell compartment and, together with microvesicles (MVs) generated by budding from the plasma membrane and apoptotic bodies, represent EVs circulating component [33]. The role of exosomes as local and systemic cell-to-cell possible mediators of oncogenic information, through horizontal transfer of various bioactive molecules, such as proteins and mRNAs, has been recently hypothesised in BC [34]. Our pilot preliminary data confirmed previously reported data [35], demonstrating that the overall blood concentration of EVs resulted significantly higher in cancer patients than in healthy volunteers. Moreover, as expected, we detected PD-L1 EVs only in the tumour, lymphoid and endothelial compartment of breast cancer patients. Preclinical studies also support the role of exosomes in TME and host immunity, and suggest exosomes as potential multi-functional therapeutic agents for TNBC [33]. Moreover, Piao et al [36], using an orthotopic TNBC model, found that TNBC-derived exosomes were able to polarize macrophages, favouring lymph node metastasis, by modifying tumour microenvironment. Recently, it has been shown that exosomes have the potential to change the fate of macrophage phenotypes, either M1, classically activated macrophages, or M2, alternatively activated macrophages on the basis of their molecular cargo [37, 38]. In terms of therapeutics, exosomes cargoes can be artificially modulated to provide pro-inflammatory, anti-tumourigenic microenvironment to suppress the tumour growth and metastasis.
The reactivation of immunosurveillance is critical for better prognosis and improved patient survival; to this purpose, it has been developed a strategy to induce ICD, a type of tumour cell death, which primes an anticancer immune response. In response to ICD, tumour cells expose calreticulin (CRT) on the cell surface prior to death and release DAMP molecules, such as adenosine triphosphate (ATP) during apoptosis or HMGB1 upon secondary necrosis, which stimulate the recruitment of DCs into the tumour bed, the uptake and processing of tumour antigens, and the antigen presentation to T cells [39]. As a consequence, the increase in the number of TILs, with high ratio CD8 /Treg, and activated CD8 , may elicit a direct cytotoxic response to eradicate cancer cells, through the generation of IFNg, perforin-1, and granzyme B [40] (Figure 1).
A lot of chemotherapeutic drugs have been used to induce ICD [39], but in the recent years, the interest has focused on oncolytic virotherapy as a strategy to stimulate ICD-hallmarks production, particularly IFN, which takes advantage of competent replicating viruses to destroy cancer cells.
Oncolytic virotherapy
The aim of oncolytic virotherapy is to stimulate anti-cancer immune response, targeting and killing tumour cells while sparing normal cells. In this perspective, natural or engineered viruses, able to transform a ‘cold’ into a ‘hot’ TME, with increased immune cell and cytokine infiltration (Figure 2). Yet, several clinical trials are evaluating the combination of oncolytic virotherapy and ICI [41]. In the clinical setting, intravenous oncolytic Orthoreovirus has been shown to increase T cell infiltration in brain tumours and to up-regulate IFN-regulated gene expression and PD-L1 expression, creating a favourable TME for subsequent ICI therapy [42]. In melanoma patients, the sequential administration of an attenuated herpes simplex virus type 1 (HVS-1), and an anti-PD-1 therapy resulted in a 33% complete response rate, with increased CD8 T cells, PDL1 protein and IFNg gene expression in responders. Baseline CD8 T cell infiltration or a baseline IFNg signature was not associated with response [43]. Oncolytic viral therapy is therefore a potential future option to skew the TME towards an ICI responsive phenotype.
Oncolytic viruses (OVs)
OVs are RNA- or DNA-attenuated viruses able to kill tumour cells after having infected them, inducing TME remodelling and antitumour immune response. Most available oncolytic viruses are genetically modified to reduce virulence for non-neoplastic host cells.
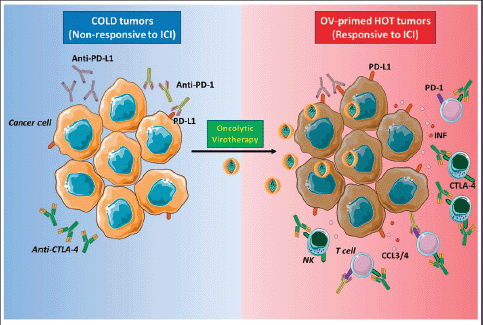
Figure 2. Oncolytic viruses make tumours ‘hot’. ‘Cold’ tumours poorly infiltrated by immune cells and have low expression of PD-L1 on cancer cells surface, and for this reason, the response to ICI therapy is inefficient (left panel). Oncolytic virotherapy promotes strong antiviral immune response accompanied by the production of cytokines, such as IFN type I that stimulates PD-L1 expression on the surface of cancer cells, and chemokines, like CCL3 and CCL4, attract immune cells expressing PD-1 or CTLA-4. These events make tumour ‘hot’, and when ICIs are administered subsequently, they can bind to their respective targets on either cancer or immune cells (right panel). NK: Natural killer; PD-1: Programmed death-1; PD-L1: Programmed death ligand-1; CTLA-4: Cytotoxic T-lymphocyte-associated protein 4; IFN I: Interferon type I; CCL3/4: Chemokine (C-C motif) ligand 3 or 4. The figure was modified from Servier Medical Art, http://smart.servier.com/.
Since 1904 many researchers have been studying the relationship between viruses and tumour cells [44, 45], focused on virus growth in tumours, rather than studying the effects of these viruses on tumour growth. The first clinical trial was reported in 1940, when an attenuated virus against melanoma led to a remarkable partial remission [46], and in 1949, the first preclinical study described the ability of the Russian Far East Virus to inhibit the growth of five transplanted mouse sarcoma 180 [47]. In 1965, Lindenmann and Klein [48] demonstrated that enhanced humoral immunity against tumour cell antigens, via the secretion of immunoglobulin and cytotoxic antibodies, was due to post-oncolytic immunity. In the next years, several viruses were studied in various leukaemia models, Hodgkins’ disease and Burkitt’s lymphoma associated with measles infection.
With the first experiences with recombinant DNA technology, in 1991, the first demonstration that herpes simplex virus (HSV) mutants (dlsptk), with depleted thymidine kinase or Infected cell protein (ICP) 34.5, were selectively replicating only in proliferating cells of human glioma xenograft was achieved [49].
To date, nine different families of viruses, including both DNA and RNA viruses, have successfully transitioned from preclinical studies to early clinical trials, with more than 570 ongoing clinical trials testing OVs [10]. This activity has now granted the approval of Talimogene Laherparepvec (T-VEC), a modified oncolytic HSV-1 for clinical use in US, Europe and Australia [50], along with the clinical use of adenovirus-derived Oncorine for head and neck cancers treatment in China [51] and native Echovirus 7 (Rigvir) for the treatment of melanoma in several European countries [52].
Mechanisms of oncolytic virus immunotherapy
OVs mediate toxic effect on tumour cells by direct or indirect mechanisms. This activity is influenced by the efficiency of cell receptor targeting, viral replication and host cell antiviral response. The lytic potential of oncolytic viruses also depends on the type of virus, dose, natural and induced viral tropism, and the susceptibility of the cancer cell to the different forms of cell death (apoptosis, necrosis, pyroptosis and autophagy) [7].
In normal cells, pathogen-associated molecular patterns (PAMPs), such as elements of viral capsids, DNA, RNA and viral protein products, active intracellular TLRs pathway, coordinate with the antiviral machinery in infected cells; this reinforces local IFN release, which, in turn, activates Protein Kinase R (PKR). PKR is a protein kinase, encoded by the EIF2AK2 gene, activated by double-stranded RNA (dsRNA) and introduced to the cells by a viral infection. It can induce cell death and viral clearance [53]. In cancer cells, IFN pathway signalling and PKR activity may be abnormal; thus, viral clearance is prevented, allowing increased viral replication. Moreover, the inherent abnormalities in the cancer cell response to stress, cell signalling and homeostasis provide a selective advantage for viral replication and in cancer cells, excluding normal cells [53]. As a consequence of oncolytic cell death, cancer cells also release viral PAMPS and additional ICD-hallmarks (DAMPs; for example, heat shock proteins, HMGB1, CRT, ATP and uric acid) and cytokines (e.g., type I IFNs, tumour necrosis factor-α (TNFα), IFNγ and interleukin‑12), which promote the maturation of antigen-presenting cells (APCs) such as DCs (Figure 3). DCs are considered a ‘bridge’ between innate and adaptive immunity and, for this reason, may serve as a carrier for some OVs (such as reovirus and measles) protecting from neutralising antibodies [54]. DCs have an important role in the initiation of immune responses, to control and eliminate viral infections. Yet, they activate the innate immune cells (such as NK) to respond to pathogens through non-specific mechanisms [55]. In particular, ‘immature’ DCs upon pathogen recognition or stimulation become ‘mature’, expressing co-stimulatory molecules (CD40 and B7), homing receptors (i.e. CCR7) and producing cytokines which induce their migration from peripheral blood to secondary lymphoid organs, where they process antigen and exhibit it via MHC class I-II, promote the expression of corresponding costimulatory molecules on T cells (CD40L, CD28) [56] and, finally, prime T cells activation and differentiation. There are different DC subsets distinguished from CD expression (CD8, CD4, CD103 and CD11b). Different DC subsets induce different T helper (Th) cell polarization [57].
Cancer cells can overcome the immune-mediated attack by modifying TME, facilitating the recruitment of immune-suppressive cells, such as TAMs and MDSCs. OVs can modify this suppressive microenvironment through a variety of mechanisms that alter the cytokine milieu and the type of immune cells within the TME potentiating systemic immunity against the cancer [58]. Moreover, oncolytic cell death can induce the release of novel cancer antigens (neo-antigens) that may have been previously hidden from the immune system, an effect already known with ICI [59, 60]. The presence of these neo-antigens is important since it may trigger an immune response and the newly generated T cell clones may be able to circulate and kill antigen-expressing cancer cells, including cancer cells that were not infected by the virus. The nearby uninfected tumour cells may be killed by the cytotoxic perforins and granzymes released by the infected tumour cells [61].
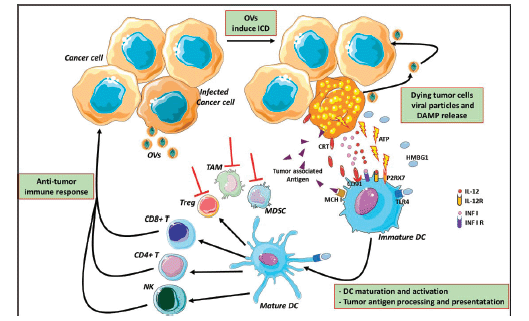
Figure 3: Mechanism of oncolytic virus immunotherapy. Oncolytic viruses infect cancer cells and induce the immunogenic cell death and release of infectious viral progeny that infect nearby cancer cells. Tumour-associated antigens and cellular DAMPs, such as CRT, HMGB1 and cellular ATP, stimulate the host antitumour immune responses. Cellular detection of viral infection and the products of oncolysis trigger the rapid activation of host antiviral responses and influx of immune cells that mediate the destruction of residual infected and uninfected tumour cells. The direct recognition and killing of tumour cells is primarily mediated by natural killer cells of the innate immune system and tumour antigen-specific CD8 cytotoxic and CD4 helper T lymphocytes of the adaptive immune system. The effects of OVs reflect on MDSCs, TAMs and Treg cells inhibition and modify the suppressive microenvironment altering the cytokine milieu (red lines) OVs: oncolytic viruses; ATP: adenosine triphosphate; HMGB1: high mobility Group Box 1; CRT: calreticulin; DAMPs: damage-associated molecular pathways; CD: cluster differentiation; TLR4: Toll-like receptor 4; P2RX7: Purin 2 Receptor X7; DC: dendritic cell; MCH I: major histocompatibility complex Class I; Treg: T regulatory; TAMs: tumour-associated microenvironment; MDSCs: myeloid-derived suppressor cells. The figure was modified from Servier Medical Art, http://smart.servier.com/.
Oncolytic virus transport and biodistribution
One of the major issues to use the OVs in therapy is the route of administration and needs to take into account physical barriers that can reduce the spread of OVs, such as the presence of necrosis, calcification, hypoxia, acidosis, increased proteolytic activity, high interstitial pressure, blood–brain barrier, tumour size and heterogeneity, dense extracellular matrix and poorly vascularisation [62]. Intratumoural (IT) injections is the main strategy used in the majority of clinical studies with OVs since it allows maximal delivery of high viral titres to tumours, bypassing systemic neutralisation and premature clearance. The main disadvantage is represented by physical inaccessibility for those tumours that are not accessible by clinical palpation; in this case, localisation via interventional imaging or surgical exposure might be required, which may be challenging for OVs repeated administrations over time. The ability of OVs to induce systemic anti-tumour response, as discussed in the previous section, could be a solution to overcome this limitation, as evidenced in the T‑VEC OPTIM phase III clinical trial [9.] In the case of Seneca Valley Virus (SVV) [63] or the chimeric adenovirus, enadenotucirev [64], delivery by intravenously (IV) administration has been shown to allow viral distribution at any site, precluding the need for additional training and interventional procedures associated with IT delivery. In the case of SVV, it can be delivered IV because of natural resistance to hemagglutination, a process resulting in premature viral clearance and reduced delivery to the tumour site following intravenous delivery [63]. The issues with IV delivery of any OVs includes considerable dilution in the systemic circulation and the likelihood of premature neutralisation through serum anti-viral immunoglobulins or other serum proteins, especially after multiple infusions. Of note, viruses naturally cause viremia and are more susceptible to antibodies neutralisation; thus, the intravenous administration of these viruses may limit the effects in patients who have had previous treatments or vaccination. For this reason, it is important to assess the viral presence not only in the tumour lesion but also in the blood. Normally, tumour specimens are tested for the presence of virus using IHC and confirmed by PCR assay. However, PCR is a highly sensitive assay for viral sequences, but have not a confirmation of live viral particles and on-going replication and more informative test are been required. In recent years, the sensitivity and accuracy of real-time PCR assays have been greatly improved by the droplet digital PCR technology (ddPCR), whose detection limit is significantly lower when compared to that of real time PCR [65].
Different approaches, such as the use of cell carriers, immunomodulators and liposomes have also been proposed. Yet, dendritic and T cells [54], mesenchymal stem cells [66] or carcinoma cells [67], could be a carrier of viruses to tumour sites, also in the presence of neutralising antibodies. The concomitant administration of immunomodulatory drugs, such as cyclophosphamide, seems to rescue OVs [68]. Clinical trials are ongoing to understand if OVs could be considered in combination or as a neoadjuvant therapy with cyclophosphamide [8]. Liposomes were also used in pre-clinical studies to encapsulate oncolytic alphavirus strain M1 (M-LPO) [69] and ONYX-015-based plasmid [70]. Liposomes harbouring the plasmids were resistant to antibodies neutralising the parent strain, while the plasmids could only transfect tumour cells which are p53 deficient [70].
Recent studies highlight the importance of MVs in cancer development. These specialised MVs are able to transfer bioactive molecules from one cell to another [33]. Tang et al [71] have demonstrated that tumour cell-derived MVs (T-MV), containing both tumour antigens and innate signals, can serve as safe and efficient carriers to deliver chemotherapeutic drugs to tumour cells. Moreover, they reported that T-Mvs can be utilised as a unique carrier system to deliver oncolytic adenoviruses to human tumours, leading to highly efficient cytolysis of tumour cells needed for in vivo treatment efficacy [72]. Exosomes are MVs (100–150 nm) that can influence viral infection in different ways, in relation to type of virus and cells. For example, exosomes isolated from human immunodeficiency virus (HIV) infected cells contained negative regulatory factors, transactivation response elements, viral microRNAs (vmiR-88, vmiR-99 and vmiR-TAR), CCR5, and CXCR4—all of which facilitate HIV-1 invasion [73]. Hepatitis C virus (HCV) utilises exosomes to transfer its genomic RNA, antisense RNA, and proteins, which aid the virus in establishing a productive infection [74]. Exosomes can also confer membranes to non-enveloped viruses, such as hepatitis A virus, allowing to escape host immune recognition. Exosomes deriving from liver nonparenchymal cells can transmit interferon (IFN)-α induced antiviral activity to hepatitis B virus replication hepatocytes [75].
Development of oncolytic viruses as drugs
The rules of a well-designed OV approach are mainly two: 1) to attenuate the viral pathogenesis and 2) to target and kill tumour cells, selectively. Natural tropism for surface molecule, altered cancer cell pathways together with the flexibility of recombinant engineering have allowed the investigation of several strategies to enhance the effectiveness of OVs. There are some molecules that are overexpressed on cancer cell surface (CD46, CD155, CD54, CD55, alfa2beta1, laminin receptor, etc.) and that can be used by OVs to recognise and entry into target cells. Some OVs, like HSV-1 and Coxsackievirus, naturally recognise the overexpressed molecules (herpesvirus entry mediator – HVEM- and CD54, respectively) and can enter in the cancer cell [76, 77]; other OVs can be engineered to directly target unique cell surface receptors, as in the case of adenovirus Ad5/3‑Δ24, which was modified to bind to integrins that are highly expressed on ovarian cancer cells [78].
A long intracellular incubation time is needed to allow OV replication and spread, explaining the preference for tumour cells infection, characterised by aberrant signalling pathways that promote resistance to cell death and proliferation. For example, Newcastle disease virus (NDV) targets B cell lymphoma (BCL) [79] or Reovirus and Vaccinia virus show natural selectivity for cells with overexpression of the RAS signalling pathway [80]. Furthermore, the possibility to insert viral genes under active promoters in cancer cells, allows limiting viral replication only in cancer cells. This is the case of a phase I trial for patients with prostate cancer, treated with adenovirus (CV706) in which the E1A, an adenoviral protein that inhibits cell cycle, was placed under promoter of prostate-specific antigen (PSA) [81]. In prostate cancer cells the promoter of PSA is highly active, E1A is selectively expressed only in these cells, resulting in proliferation of adenovirus and virus mediated cell lysis [81].
Since normal and cancer cells exhibit differential expressions of cognate miRNA elements, the engineered OVs with miRNA Target Sequencing (miRTA) can be blocked from replicating in normal cells, where specific miRNAs are expressed. This type of construct was designed in glioma, where it showed limited viral replication in neurons that constitutively express high levels of miR‑7, while allowing proliferation in glioma cells, that frequently downregulate miR‑7 [82]. Recent evidence has demonstrated that exosomal miRNA can not only be secreted by cancer cells but it can also be uptake by these cells. It has been also hypothesised that NDV might use exosomes to entry into neighbouring miRNA carrying cells, resulting in inhibition of the IFN pathway and promotion of viral infection [83].
Oncolytic virus therapy in breast cancer
In recent years, the role of ICI has been extensively studied in breast cancer patients, with particular emphasis on triple negative disease leading to FDA and EMA approval of atezolizumab in the first line treatment of PD-L1 overexpressing TN MBC [5]. Moreover, extensive research is ongoing to characterise the prognostic role of TILs [84]. In this perspective, a powerful strategy to enhance the response to ICI, such as anti-PD1/PD-L1, is TME reprogramming: yet, the administration of chemotherapy agents, such as doxorubicin and cisplatin, has been shown to induce up-regulation of inflammatory JAK-STAT and TNF-α signalling and to increase responsiveness to Nivolumab in metastatic TNBC [85]. Innovative treatment options, such as OVs, are studied as a monotherapy or in combination with ICI or CT, both in pre-clinical and clinical trials.
Oncolytic Herpes Simplex Virus (HSV)
HSV‑1 is a double-stranded DNA virus with a large genome (152 kb), member of the alphaherpesvirus family; it replicates in the nucleus and is not able to induce insertional mutagenesis, making HSV‑1 an attractive candidate for therapeutic development. Human breast cancer cells are permissive to HSV-1 and genetic manipulations allow virus selectivity, while sparing normal cells [86].
Talimogene laherparepvec (T‑VEC) is the most commercially advanced HSV‑1‑ based OV. T‑VEC contains deletions of two genes: ICP34.5 and ICP47. ICP34.5 is a neurovirulence gene, critical for blocking the host antiviral PKR–IFN response; its deletion confers cancer selectivity and prevention of neural infection. The two ICP34.5 genes have been substituted with the gene encoding granulocyte-macrophage colony-stimulating factor (GM-CSF) to improve the induction of antitumour immunity. ICP47 deletion results into viral antigen presentation, leading to the containment of the infection in healthy tissues and immune-mediated kill of cancer cells that selectively propagate oncolytic HSV‑1. The deletion of ICP47 also induces the early activation of the US11 promoter that blocks PKR phosphorylation preventing cancer cell apoptosis [86].
In preclinical studies, T‑VEC demonstrated potent lytic effects against several tumour cell lines, most notably melanoma and pancreatic cancer cells [10]. T‑VEC has been evaluated in clinical trials in patients with melanoma, pancreatic cancer, head and neck tumours, and in patients with breast cancer in a phase I clinical trial [87]. Treatment was generally well tolerated, with most adverse events related to fever, chills, nausea, fatigue and local injection site reactions.
T-VEC may represent a valuable tool in combination with ICIs, particularly in those tumours with a low baseline lymphocyte infiltrate that are poorly responsive to ICI as a single agent, such as breast cancer. A phase 1b clinical trial, including 21 advanced melanoma patients treated with T-VEC followed by pembrolizumab, showed an interesting level of activity of the anti-PD-1 agent, related to TME modulation [43]. The safety and efficacy of T-VEC (as a monotherapy or combination therapy with paclitaxel) in TNBC patients is under evaluation in a clinical trial [17]. Recently, antitumour activity of G47Δ-mIL12 was observed in a 4T1 tumour model of advanced breast cancer. G47Δ-mIL12 (14) is a genetically engineered oncolytic HSV (oHSV) that has similar genetic modifications to T-VEC but contains ICP6 inactivation, limiting HSV replication to cancer cells and expressing murine Interleukin 12 (IL-12) (instead of GM-CSF). The G47Δ-mIL12 efficacy has been tested both in vitro and in vivo experiments. In vitro, it was able to infect and eliminate both murine and human TNBC cells. In vivo, G47Δ-mIL12 treatment effectively inhibited 4T1 tumour growth in a CD8 T cell-dependent fashion, and prevented metastatic spread [88].
Administration of HF10 in immunocompetent mouse models of colon and breast cancer stimulated specific antitumour immune responses and provided resistance to malignant cell re-implants [89]. HF10 is a spontaneously mutated HSV-1 and is expected to be a promising agent in combination with bevacizumab, since bevacizumab enhances viral distribution, increases tumour hypoxia and expands the population of apoptotic cells, therefore producing a synergistic antitumour effect [90]. An investigator-initiated phase I trial of HF10 enrolled six patients with cutaneous and subcutaneous metastatic breast cancer. A histological examination revealed fibrosis and tumour cell death with an infiltration of CD8 and CD4 T-cells around tumour islets, supporting the induction of an immune response [91].
Oncolytic adenovirus
Adenoviruses are medium-sized (70–90 nm), non-enveloped icosahedral viruses with double-stranded DNA. Many types of immunologically distinct adenoviruses can cause infections in humans. Adenovirus expresses coxsackie-adenovirus receptor (CAR) and adenoviral early genes (encoded by E1A and E1B) that allow the cell infection targeting suppressors oncogene p53 and retinoblastoma-associated protein (pRb). Normal cells are protected by p53 and pRb activities, whereas in cancer cells these genes are compromised, and adenovirus can interfere with cell cycle proteins. Some of the strategies for modification of adenovirus are described, as attenuation, and the use of oncolytic adenovirus in clinical practice has registered very few adverse events.
Oncolytic adenovirus provides a novel therapeutic modality for breast cancer with its appropriate targeting property. Adenoviruses ONYX‑015, which has a deletion in the portion of E1B that inactivates p53, are currently investigated in a phase I trial in combination with etanercept (a recombinant dimer of the human TNF); so far, this study enrolled two patients with metastatic breast cancer, as well as other patients with different types of cancer [92]. Recent findings demonstrated that a novel recombinant adenovirus, targeting E2F-1 and IL-15, has an inhibitive effect on breast cancer proliferation [93]. The transcriptional factor E2F-1 plays a significant role in the control of cell cycle, proliferation and carcinogenesis, and it is higher expressed in breast cancer tissues compared with normal tissues, suggesting that E2F-1 may be an effective target for treatment. Also, by carrying human Interleukin-15 (IL-15) gene, a crucial cytokine enhancing the activity of macrophages and neutrophils, it may induce the production and activation of effector T cells and regulate the survival and proliferation of memory T cells, giving to the oncolytic adenovirus an immunomodulatory effect [93]. Other pre-clinical studies highlighted the importance of oncolytic adenoviruses engineered interfering lncRNA [94] or targeting TGF-β in cancer cells [95].
Pelareorep (Reolysin®)
Reolysin (Oncolytics Biotech Inc., Calgary, Alberta, Canada) is a purified live replication-competent form of the reovirus serotype 3 Dearing strain. REOvirus (Respiratory Enteric Orphan), belonging to the genus Orthoreovirus, family Reoviridae, is a 70 nm, naturally occurring, ubiquitous, non-enveloped, an icosahedral shaped virus with a genome of 10 segments of double-stranded RNA. The application of reovirus as an oncolytic virus exploits the RAS pathway that is linked to the activation of PKR [96]. A phase II, randomised, clinical trial of pelareorep in combination with paclitaxel in patients with metastatic breast cancer has shown that this combination is more effective than paclitaxel alone [97]. Recently, in a pre-clinical study, it has been demonstrated that reovirus was able to induce an immune response against breast cancer cells when combined with anti-PD-1 therapy [98].
Vaccinia virus
Vaccinia virus is a dsDNA virus and is a member of the poxvirus family; they can infect a wide range of cells entering by endocytic mechanism and replicating in the cytosol. For its large capacity of transgenes, easy manipulation, a good safety profile, and the inability to integrate into the host genome, the vaccinia virus is considered a good OV. Specifically, viral thymidine kinase, vaccinia growth factor and vaccinia type I IFN-binding protein (B18R) have been modified to increase cancer cell selectivity and lysis [99]. Based on the importance of the immune response and the ability of the vaccinia viral genome to accept large transgenes (25 kb), vaccinia virus has been engineered to express tumour antigens, T cell co‑stimulatory molecules and inflammatory cytokines [7]. Recently, an engineered oncolytic vaccinia virus ((VV)-iPDL1/GM) that co-expresses a PD-L1 inhibitor and GMCSF has been developed. This OV can secrete the PD-L1 inhibitor, that systemically binds and inhibits PD-L1 on tumour cells and immune cells but can activate tumour neoantigen-specific T cell responses, via GM-CFS stimulation. This provides a tumour-specific oncolytic immunotherapy for cancer patients, especially those resistant to PD-1/PD-L1 blockade therapy [100].
Moreover, the Western Reserve strain of vaccinia virus (VV), the most virulent strain of VV in animal models, has been engineered for tumour selectivity through two targeted gene deletions (vvDD). vvDD were administered via IT dose escalation in 16 patients with advanced solid tumours (first-in-human phase 1). The study report that it was well-tolerated, showing a selective antitumour activity [101].
Newcastle disease virus
NDV is a single-stranded RNA enveloped avian paramyxovirus that ranges in size from 100 to 500 nm. NDV infects cells through plasma membrane fusion or through direct endocytosis of the virus. Cancer cell specificity is conferred by its sensitivity to type I IFNs and overexpression of BCL-XL. NDV induces apoptosis and directly activates the innate immune system through increased cytokine production (type I IFN, RANTES, IL‑12, and GM‑CSF) and improved antigen presentation. The NDV protein haemagglutinin-neuraminidase can act as a potent antigen that augments cytolytic T cell responses against infected cells. NDV-induced apoptosis results in the conversion of an immune-suppressive tumour microenvironment into a pro-inflammatory environment that supports antitumour immune responses.
NDV infection usually has minimal clinical symptoms, making this virus optimal for cancer treatment. This is due to the fact that cancer cells are relatively permissive to NDV infection. In vitro and in vivo data have shown the effectiveness of different strains of oncolytic NDV as novel strategy for breast cancer therapy. A newly developed recombinant NDV that expresses IL12 (rAF-IL12) has been observed to inhibit tumour growth significantly (52%) in treated mice [102]. Recently, it has been demonstrated that combining NDV virotherapy with glucose analogue 2-deoxyglucose (2-DG), a enhances the anti-tumour effect in human- and mouse-breast cancer cells [103]. These results open the possibility to apply the use of OVs and in particular NDV in different strategies to treat breast cancer. The encapsulation of NDV in exosomes has been proposed as an innovative approach for virus delivery. Several studies have shown that exosomes are involved in viral infections and in intercellular communication, through miRNA. In a recent study performed in HeLa cell line, it has been speculated that NDV used exosomes to entry into neighbouring cells, the so-called ‘recipient cell’, carrying miRNAs produced by the original cell, called the ‘parent cell’, after virus infection; this results in IFN pathway inhibition and promotion of viral infection of the ‘recipient cell’ [104].
Although numerous preclinical studies have suggested that NDV has antitumour activity against a wide variety of cancers, there are only a limited number trials in progress at present [105]. The effect of NDV (PV701) has been evaluated in two BC patients enrolled into a phase I trial. PV701 was IV injected twice at lower dosages followed by dose escalation. The treatment regimen was well tolerated [106].
Other OVs
There are other viruses under investigation for their oncolytic activity both as a single agent as well as in combination with other anticancer agent: coxsackievirus, poliovirus, vesicular stomatitis virus (VSV), measles virus and Maraba virus.
In particular, coxsackievirus strains and poliovirus reported promising results, both in vivo and in vitro, with favourable toxicity profile [107–109].
Oncolytic measles viruses (oMV) derived from the attenuated Edmonston-B vaccine strain can recognise target cells through overexpression of specific molecules such as CD46, signalling lymphocytic activation molecule, the poliovirus receptor-related 4 protein and Nectin Cell Adhesion Molecule 4 (Nectin 4). Nectin 4 levels can be downregulated by specific miRNA (i.e., miR-31 and miR-128) with the impact on oMV infectivity in vitro and in xenograft models [110]. It has also been observed that the Aurora A kinase inhibitor alisertib enhanced oMV oncolysis in vitro and improved outcome in vivo against breast cancer xenografts [111]. Intriguingly, a group of researchers demonstrated that, using dendritic cells as a carrier for oMV, it is possible to overcome the problem of antibody neutralisation with promising results in terms of virus delivery to breast cancer cells [112]. To date there is one phase I clinical trial that is currently open (NCT01846091), whose primary aim is to evaluate the toxicity of this approach in MBC.
Oncolytic rhabdoviruses like Maraba and VSV have been shown to enhance NK cell–mediated killing of tumour cells, dendritic cell maturation, up-regulation of antigen presentation by tumour cells and production of proinflammatory cytokines and chemokines [113]. Maraba rhabdovirus has been tested in a variety of mouse tumour models [113] and is now being evaluated in phase 1/2 studies (NCT02285816 and NCT02879760).
Conclusion
Viruses used as a weapon against cancer is one the revolutionary discoveries of recent years.
The two main advantages in the use of oncolytic virotherapy are: 1) its specificity for tumour cells and 2) its active role in triggering anti-tumour immunity already present, as in the case of ‘hot’ tumours, and that of systemic immune cells attracted in TME, in ‘cold’ tumours. However, if, on the one hand, this form of immunomodulation is characterised by moderate toxicity, increased specificity, and hopefully activity, some issues must be optimised in the development of OVs as a new class of drugs. In fact, appropriate dosages and schedules, pharmacodynamic assays, delivery systems and type of clinical trial designs need to be carefully assessed, especially in combination regimens with CT or ICIs, to improve performance and results. Furthermore, intermediate endpoints, such as predictive biomarkers. Patient selection must also be carefully evaluated in terms of immune system performance.
Oncolytic virotherapy is located in the context of personalised medicine, since its best application is associated to the interplay between human immune system and TME. Many clinical trials including oncolytic virotherapy are phase II/III trials (Table 1), due to the need to increase the knowledge on specific mechanisms between OVs, human immune system and tumour, mainly to prevent adverse events. Severe adverse events caused by oncolytic virotherapy have rarely been reported, while moderate adverse events can be controlled or disappear spontaneously, avoiding viremia. Today, the OVs are administrated via IT and IV delivery, although the most appropriate route of administration has not yet been defined, to reduce adverse events and enhance OVs efficacy. In clinical trials, IV delivery may be preferable to IT injection: this approach, in fact, simplifies the interventional procedures and improves systemic delivery to distant metastasis. However, IV administration is associated with excessive dilution and premature neutralisation through serum anti-viral immunoglobulins or other serum proteins. To overcome this, it is recommended to use rapid infusion schedules, with repeated high doses to avoid serum neutralising antibodies, and preferentially in combination with other anticancer therapies.
The development of new oncolytic virus for the treatment of breast cancer includes new viral vectors targeting tumour cells with high affinity without perturbating normal cells, improving immune system activity against cancer, reducing adverse events and ensuring safety. In particular, in breast cancer, novel reovirus, vaccinia virus and HSV are been developing. Among these, vaccinia virus is considered to be very promising candidate, due to its transfection capacity, easy manipulation, good safety profile and inability to integrate into the host genome.
At present, there are several phase I and II clinical trials using OVs to treat breast cancer patients (Table 1); most of these use a combination approach. On the other hand, a number of preclinical trials are studying alternative combination approaches concerning delivery systems and dosages. In conclusion, oncolytic virotherapy seems to show good promise for breast cancer therapy, with potentially effective and well-tolerated regimens, and possibly improving quality of life.
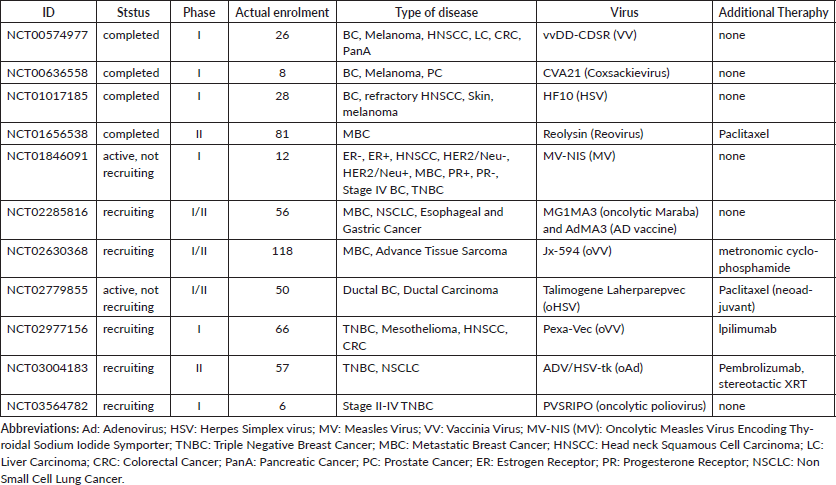
Table 1: Clinical Trials for breast cancer treatment using oncolytic virotherapy.
Acknowledgments
This work was supported by the Italian Association for Cancer Research and AGING Project – Department of Excellence – DIMET, Università del Piemonte Orientale, Novara, Italy.
Conflicts of interest
There are no conflicts of interest.
References
1. Chen DS and Mellman I (2013) Oncology meets immunology: the cancer-immunity cycle Immunity 39(1) 1–10 https://doi.org/10.1016/j.immuni.2013.07.012 PMID: 23890059
2. Kyi C and Postow MA (2014) Checkpoint blocking antibodies in cancer immunotherapy FEBS Lett 588(2) 368–376 https://doi.org/10.1016/j.febslet.2013.10.015
3. Darvin P, Toor SM, and Sasidharan Nair V, et al (2018) Immune checkpoint inhibitors: recent progress and potential biomarkers Exp Mol Med 50(12) 1–11 https://doi.org/10.1038/s12276-018-0191-1 PMID: 30546008 PMCID: 6292890
4. Hegde PS, Karanikas V, and Evers S (2016) The where, the when, and the how of immune monitoring for cancer immunotherapies in the era of checkpoint inhibition Clin Cancer Res 22(8) 1865–1874 https://doi.org/10.1158/1078-0432.CCR-15-1507 PMID: 27084740
5. Kok M, Voorwerk L, and Horlings H, et al (2018) Adaptive phase II randomized trial of nivolumab after induction treatment in triple negative breast cancer (TONIC trial): final response data stage I and first translational data J Clin Oncol 36(15) https://doi.org/10.1200/JCO.2018.36.15_suppl.1012
6. Mellman I, Coukos G, and Dranoff G (2011) Cancer immunotherapy comes of age Nature 480(7378) 480–489 https://doi.org/10.1038/nature10673 PMID: 22193102 PMCID: 3967235
7. Kaufman HL, Kohlhapp FJ, and Zloza A (2015) Oncolytic viruses: a new class of immunotherapy drugs Nat Rev Drug Discov 14(9) 642–662 https://doi.org/10.1038/nrd4663 PMID: 26323545 PMCID: 7097180
8. Raja J, Ludwig JM, and Gettinger SN, et al (2018) Oncolytic virus immunotherapy: future prospects for oncology J Immunother Cancer 6(1) 140 https://doi.org/10.1186/s40425-018-0458-z PMID: 30514385 PMCID: 6280382
9. Andtbacka RHI, Kaufman HL, and Collichio F, et al (2015) Talimogene laherparepvec improves durable response rate in patients with advanced melanoma J Clin Oncol 33(25) 2780–2788 https://doi.org/10.1200/JCO.2014.58.3377 PMID: 26014293
10. Peters C, Grandi P, and Nigim F (2019) Updates on oncolytic virus immunotherapy for cancers Mol Ther Oncolytics 12 259–262 https://doi.org/10.1016/j.omto.2019.01.008 PMID: 33072862 PMCID: 7536361
11. Restifo NP, Smyth MJ and Snyder A (2016) Acquired resistance to immunotherapy and future challenges Nat Rev Cancer 16(2) 121–126 https://doi.org/10.1038/nrc.2016.2 PMID: 26822578 PMCID: 6330026
12. Antohe M, Nedelcu RI, and Nichita L, et al (2019) Tumor infiltrating lymphocytes: the regulator of melanoma evolution (Review) Oncol Lett 17(5) 4155–4161 PMID: 30944610 PMCID: 6444298
13. Dong ZY, Wu SP, and Liao RQ, et al (2016) Potential biomarker for checkpoint blockade immunotherapy and treatment strategy Tumor Biol 37(4) 4251–4261 PMID: 26779629
14. García-Romo GS, García-Castillo KG, and Díaz-Rodríguez Á, et al (2017) Main immunoregulatory mechanisms favoring the development of breast cancer Gac Med Mex 153(2) 229 PMID: 28474709
15. Takeuchi Y and Nishikawa H (2016) Roles of regulatory T cells in cancer immunity Int Immunol 28(8) 401–409 https://doi.org/10.1093/intimm/dxw025 PMID: 27160722 PMCID: 4986235
16. DeNardo DG and Coussens LM (2007) Inflammation and breast cancer. Balancing immune response: crosstalk between adaptive and innate immune cells during breast cancer progression Breast Cancer Res 9(4) 212 https://doi.org/10.1186/bcr1746 PMID: 17705880 PMCID: 2206719
17. Marra A, Viale G and Curigliano G (2019) Recent advances in triple negative breast cancer: the immunotherapy era BMC Med 17 https://doi.org/10.1186/s12916-019-1326-5 PMID: 31068190 PMCID: 6507064
18. Miyashita M, Sasano H, and Tamaki K, et al (2015) Prognostic significance of tumor-infiltrating CD8 and FOXP3 lymphocytes in residual tumors and alterations in these parameters after neoadjuvant chemotherapy in triple-negative breast cancer: a retrospective multicenter study Breast Cancer Res 17(1) 124 https://doi.org/10.1186/s13058-015-0632-x PMID: 26341640 PMCID: 4560879
19. Cho HJ, Jung JI, and Lim DY, et al (2012) Bone marrow-derived, alternatively activated macrophages enhance solid tumor growth and lung metastasis of mammary carcinoma cells in a Balb/C mouse orthotopic model Breast Cancer Res 14(3) R81 https://doi.org/10.1186/bcr3195 PMID: 22616919 PMCID: 3446344
20. Green DR, Ferguson T, Zitvogel L, et al (2009) Immunogenic and tolerogenic cell death Nat Rev Immunol 9(5) 353 https://doi.org/10.1038/nri2545 PMID: 19365408 PMCID: 2818721
21. Bezu L, Gomes-de-Silva LC, and Dewitte H, et al (2015) Combinatorial strategies for the induction of immunogenic cell death Front Immunol 6 187 https://doi.org/10.3389/fimmu.2015.00187 PMID: 25964783 PMCID: 4408862
22. McCracken MN, Cha AC and Weissman IL (2015) Molecular pathways: activating T cells after cancer cell phagocytosis from blockade of CD47 ‘Don’t eat Me’ signals Clin Cancer Res 21(6) 3597–3601 https://doi.org/10.1158/1078-0432.CCR-14-2520 PMID: 26116271 PMCID: 4621226
23. Willingham SB, Volkmer JP, and Gentles AJ, et al (2012) The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors Proc Natl Acad Sci 109(17) 6662–6667 https://doi.org/10.1073/pnas.1121623109 PMID: 22451913 PMCID: 3340046
24. Gholamin S, Mitra SS, and Feroze AH, et al (2017) Disrupting the CD47-SIRPα anti-phagocytic axis by a humanized anti-CD47 antibody is an efficacious treatment for malignant pediatric brain tumors Sci Transl Med 9(381) eaaf2968 https://doi.org/10.1126/scitranslmed.aaf2968 PMID: 28298418
25. Kaur S, Elkahloun AG, and Singh SP, et al (2016) A function-blocking CD47 antibody suppresses stem cell and EGF signaling in triple-negative breast cancer Oncotarget 7(9) 10133–10152 https://doi.org/10.18632/oncotarget.7100 PMID: 26840086 PMCID: 4891109
26. Tariq M, Zhang J, and Liang G, et al (2017) Macrophage polarization: anti-cancer strategies to target tumor-associated macrophage in breast cancer J Cell Biochem 118(9) 2484–2501 https://doi.org/10.1002/jcb.25895 PMID: 28106295
27. Allavena P, Sica A, and Solinas G, et al (2008) The inflammatory micro-environment in tumor progression: the role of tumor-associated macrophages Crit Rev Oncol Hematol 66(1) 1–9 https://doi.org/10.1016/j.critrevonc.2007.07.004
28. Bronte V, Brandau S, and Chen S-H, et al (2016) Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards Nat Commun 7 https://doi.org/10.1038/ncomms12150 PMID: 27381735 PMCID: 4935811
29. Marvel D and Gabrilovich DI (2015) Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected J Clin Investig 125(9) 3356–3364 https://doi.org/10.1172/JCI80005 PMID: 26168215 PMCID: 4588239
30. Vincent J, Mignot G, and Chalmin F, et al (2010) 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity Cancer Res 70(8) 3052–3061 https://doi.org/10.1158/0008-5472.CAN-09-3690 PMID: 20388795
31. Shirota Y, Shirota H and Klinman DM (2012) Intratumoral injection of CpG oligonucleotides induces the differentiation and reduces the immunosuppressive activity of myeloid-derived suppressor cells J Immunol 188(4) 1592–1599 https://doi.org/10.4049/jimmunol.1101304 PMID: 22231700 PMCID: 3273593
32. Ozawa PMM, Alkhilaiwi F, and Cavalli IJ (2018) Extracellular vesicles from triple-negative breast cancer cells promote proliferation and drug resistance in non-tumorigenic breast cells Breast Cancer Res Treat 172(3) 713–723 https://doi.org/10.1007/s10549-018-4925-5 PMID: 30173296 PMCID: 6245099
33. György B, Szabó TG, and Pásztói M, et al (2011) Membrane vesicles, current state-of-the-art: emerging role of extracellular vesicles Cell Mol Life Sci 68(16) 2667–2688 https://doi.org/10.1007/s00018-011-0689-3 PMID: 21560073 PMCID: 3142546
34. Tutanov O, Orlova E, and Proskura K, et al (2020) Proteomic analysis of blood exosomes from healthy females and breast cancer patients reveals an association between different exosomal bioactivity on non-tumorigenic epithelial cell and breast cancer cell migration in vitro Biomolecules 10(4) 495 https://doi.org/10.3390/biom10040495 PMCID: 7226042
35. Brocco D, Lanuti P, and Simeone P, et al (2019) Circulating cancer stem cell-derived extracellular vesicles as a novel biomarker for clinical outcome evaluation J Oncol 2019 https://doi.org/10.1155/2019/5879616 PMID: 31827511 PMCID: 6885781
36. Piao YJ, Kim HS, and Hwang EH, et al (2018) Breast cancer cell-derived exosomes and macrophage polarization are associated with lymph node metastasis Oncotarget 9(7) 7398–7410 https://doi.org/10.18632/oncotarget.23238 PMID: 29484119 PMCID: 5800911
37. Park JE, Dutta B, and Tse SW, et al (2019) Hypoxia-induced tumor exosomes promote M2-like macrophage polarization of infiltrating myeloid cells and microRNA-mediated metabolic shift Oncogene 38(26) 5158–5173 https://doi.org/10.1038/s41388-019-0782-x PMID: 30872795
38. Bardi GT, Smith MA, and Hood JL (2018) Melanoma exosomes promote mixed M1 and M2 macrophage polarization Cytokine 105 63–72 https://doi.org/10.1016/j.cyto.2018.02.002 PMID: 29459345 PMCID: 5857255
39. Wang YJ, Fletcher R, and Yu J, et al (2018) Immunogenic effects of chemotherapy-induced tumor cell death Genes Dis 5(3) 194–203 https://doi.org/10.1016/j.gendis.2018.05.003 PMID: 30320184 PMCID: 6176216
40. West NR, Milne K, and Truong PT, et al (2011) Tumor-infiltrating lymphocytes predict response to anthracycline-based chemotherapy in estrogen receptor-negative breast cancer Breast Cancer Res 13(6) R126 https://doi.org/10.1186/bcr3072 PMID: 22151962 PMCID: 3326568
41. Zamarin D, Holmgaard RB, and Subudhi SK, et al (2014) Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy Sci Transl Med 6(226) 226ra32 https://doi.org/10.1126/scitranslmed.3008095 PMID: 24598590 PMCID: 4106918
42. Samson A, Scott KJ, and Taggart D, et al (2018) Intravenous delivery of oncolytic reovirus to brain tumor patients immunologically primes for subsequent checkpoint blockade Sci Transl Med 10(422) eaam7577 https://doi.org/10.1126/scitranslmed.aam7577 PMID: 29298869 PMCID: 6276984
43. Ribas A, Dummer R, and Puzanov I, et al (2017) Oncolytic virotherapy promotes intratumoral T cell infiltration and improves anti-PD-1 immunotherapy Cell 170(6) 1109–1119 https://doi.org/10.1016/j.cell.2017.08.027 PMID: 28886381
44. Dock G (1994) The influence of complicating diseases upon leukæmia Am J Med Sci https://doi.org/10.1097/00000441-190412740-00001
45. Rivers TM and Pearce L (1925) Growth and persistence of filterable viruses in a transplantable rabbit neoplasm J Exp Med 42(4) 523–537 https://doi.org/10.1084/jem.42.4.523 PMID: 19869072 PMCID: 2131027
46. Pack GT (1950) Note on the experimental use of rabies vaccine for melanomatosis Arch Derm Syphilol 62(5) 694–695 https://doi.org/10.1001/archderm.1950.01530180083015
47. Moore AE (1949) The destructive effect of the virus of russian far east encephalitis on the transplantable mouse sarcoma 180 Cancer 2(3) 525–534 https://doi.org/10.1002/1097-0142(194905)2:3<525::AID-CNCR2820020317>3.0.CO;2-O PMID: 18131412
48. Lindenmann J, Klein PA, and Lindenmann J, et al (1967) Tumor immunity following viral oncolysis Immunological Aspects of Viral Oncolysis https://doi.org/10.1007/978-3-642-87044-6_5
49. Martuza RL, Malick A, and Markert JM, et al (1991) Experimental therapy of human glioma by means of a genetically engineered virus mutant Science (80) https://doi.org/10.1126/science.1851332 PMID: 1851332
50. Greig SL (2016) Talimogene laherparepvec: first global approval Drugs 76(1) 147–154 [http://rug.on.worldcat.org/atoztitles/link/?sid=EMBASE&issn=11791950&id=doi:10.1007/s40265-015-0522-7&atitle=Talimogene Laherparepvec: First Global Approval&stitle=Drugs&title=Drugs&volume=76&issue=1&spage=147&epage=154&aulast=Greig&aufirst=Sarah L.&auinit=S.L.&aufull=Greig S.L.&coden=DRUGA&isbn=&pages=147-154&date=2016&auinit1=S&auinitm=L] https://doi.org/10.1007/s40265-015-0522-7
51. Garber K (2006) China approves world’s first oncolytic virus therapy for cancer treatment J Natl Cancer Inst 98(5) 298–300 https://doi.org/10.1093/jnci/djj111 PMID: 16507823
52. Alberts P, Tilgase A, and Rasa A, et al (2018) The advent of oncolytic virotherapy in oncology: the Rigvir® story Eur J Pharmacol 837 117–126 https://doi.org/10.1016/j.ejphar.2018.08.042 PMID: 30179611
53. Elde NC, Child SJ, and Geballe AP, et al (2009) Protein kinase R reveals an evolutionary model for defeating viral mimicry Nature 457(7228) 485–489 https://doi.org/10.1038/nature07529 PMCID: 2629804
54. Ilett EJ, Prestwich RJ, and Kottke T, et al (2009) Dendritic cells and T cells deliver oncolytic reovirus for tumour killing despite pre-existing anti-viral immunity Gene Ther 16(5) 689–699 https://doi.org/10.1038/gt.2009.29 PMID: 19282847 PMCID: 4562026
55. Freer G and Matteucci D (2009) Influence of dendritic cells on viral pathogenicity PLoS Pathogens https://doi.org/10.1371/journal.ppat.1000384 PMID: 19649323 PMCID: 2712770
56. Steinman RM (2007) Lasker basic medical research award. Dendritic cells: versatile controllers of the immune system Nat Med 13(10) 1155–1159 https://doi.org/10.1038/nm1643 PMID: 17917664
57. Kapsenberg ML (2003) Dendritic-cell control of pathogen-driven T-cell polarization Nat Rev Immunol 3(12) 984–993 https://doi.org/10.1038/nri1246 PMID: 14647480
58. Bridle BW, Stephenson KB, and Boudreau JE, et al (2010) Potentiating cancer immunotherapy using an oncolytic virus Mol Ther 18(8) 1430–1439 https://doi.org/10.1038/mt.2010.98 PMID: 20551919 PMCID: 2927075
59. Kanerva A, Nokisalmi P, and Diaconu I, et al (2013) Antiviral and antitumor T-cell immunity in patients treated with GM-CSF-coding oncolytic adenovirus Clin Cancer Res 19(10) 2734–2744 https://doi.org/10.1158/1078-0432.CCR-12-2546 PMID: 23493351
60. Snyder A, Makarov V, and Merghoub T, et al (2014) Genetic basis for clinical response to CTLA-4 blockade in melanoma N Engl J Med 371(23) 2189–2199 https://doi.org/10.1056/NEJMoa1406498 PMID: 25409260 PMCID: 4315319
61. Schietinger A, Philip M, and Liu RB, et al (2010) Bystander killing of cancer requires the cooperation of CD4 and CD8 T cells during the effector phase J Exp Med 207(11) 2469–2477 https://doi.org/10.1084/jem.20092450 PMID: 20921286 PMCID: 2964573
62. Nguyen A, Ho L and Wan Y (2014) Chemotherapy and oncolytic virotherapy: Advanced tactics in the war against cancer Front Oncol 4 145 https://doi.org/10.3389/fonc.2014.00145 PMID: 24967214 PMCID: 4052116
63. Rudin CM, Poirier JT, and Senzer NN, et al (2011) Phase I clinical study of Seneca Valley Virus (SVV-001), a replication-competent picornavirus, in advanced solid tumors with neuroendocrine features Clin Cancer Res 17(4) 888–895 https://doi.org/10.1158/1078-0432.CCR-10-1706 PMID: 21304001 PMCID: 5317273
64. Machiels JP, Salazar R, and Rottey S, et al (2019) A phase 1 dose escalation study of the oncolytic adenovirus enadenotucirev, administered intravenously to patients with epithelial solid tumors (EVOLVE) J Immunother Cancer 7 https://doi.org/10.1186/s40425-019-0510-7
65. Olmedillas-López S, García-Arranz M and García-Olmo D (2017) Current and emerging applications of droplet digital PCR in oncology Mol Diagn Ther 21(5) 493–510 https://doi.org/10.1007/s40291-017-0278-8 PMID: 28477149
66. Pereboeva L (2003) Approaches to utilize mesenchymal progenitor cells as cellular vehicles Stem Cells 21(4) 389–404 https://doi.org/10.1634/stemcells.21-4-389 PMID: 12832693
67. Power AT, Wang J, and Falls TJ, et al (2007) Carrier cell-based delivery of an oncolytic virus circumvents antiviral immunity Mol Ther 15(1) 123–130 https://doi.org/10.1038/sj.mt.6300039
68. Qiao J, Wang H, and Kottke T, et al (2008) Cyclophosphamide facilitates antitumor efficacy against subcutaneous tumors following intravenous delivery of reovirus Clin Cancer Res https://doi.org/10.1158/1078-0432.CCR-07-1510
69. Wang Y, Huang H, and Zou H, et al (2019) Liposome encapsulation of oncolytic virus M1 to reduce immunogenicity and immune clearance in vivo Mol Pharm https://doi.org/10.1021/acs.molpharmaceut.8b01046
70. Yotnda P, Davis AR, and Hicks MJ, et al (2004) Liposomal enhancement of the antitumor activity of conditionally replication-competent adenoviral plasmids Mol Ther 9(4) 489–495 https://doi.org/10.1016/j.ymthe.2004.01.018 PMID: 15093179
71. Tang K, Zhang Y, and Zhang H, et al (2012) Delivery of chemotherapeutic drugs in tumour cell-derived microparticles Nat Commun 3 1282 https://doi.org/10.1038/ncomms2282 PMID: 23250412
72. Ran L, Tan X, and Li Y, et al (2016) Delivery of oncolytic adenovirus into the nucleus of tumorigenic cells by tumor microparticles for virotherapy Biomaterials 89 56–66 https://doi.org/10.1016/j.biomaterials.2016.02.025 PMID: 26950165
73. Sampey GC, Saifuddin M, and Schwab A, et al (2016) Exosomes from HIV-1-infected cells stimulate production of pro-inflammatory cytokines through trans-activating response (TAR) RNA J Biol Chem https://doi.org/10.1074/jbc.M115.662171 PMCID: 4714213
74. Ramakrishnaiah V, Thumann C, and Fofana I, et al (2013) Exosome-mediated transmission of hepatitis C virus Proc Natl Acad Sci USA 110(32) 13109–13113 https://doi.org/10.1073/pnas.1221899110 PMID: 23878230 PMCID: 3740869
75. Wang J, Zheng Y and Zhao M (2017) Exosome-based cancer therapy: implication for targeting cancer stem cells Front Pharmacol 7 533 https://doi.org/10.3389/fphar.2016.00533 PMID: 28127287 PMCID: 5226951
76. Shafren DR, Au GG, and Nguyen T, et al (2004) Systemic therapy of malignant human melanoma tumors by a common cold-producing enterovirus, coxsackievirus A21 Clin Cancer Res 10 53–60 https://doi.org/10.1158/1078-0432.CCR-0690-3 PMID: 14734451
77. Yu Z, Chan MK, and O-charoenrat P, et al (2005) Enhanced nectin-1 expression and herpes oncolytic sensitivity in highly migratory and invasive carcinoma Clin Cancer Res 11(3) 4889–4897 https://doi.org/10.1158/1078-0432.CCR-05-0309 PMID: 16000587
78. You Z, Fischer DC, and Tong X, et al (2001) Coxsackievirus-adenovirus receptor expression in ovarian cancer cell lines is associated with increased adenovirus transduction efficiency and transgene expression Cancer Gene Ther 8(3) 168–175 https://doi.org/10.1038/sj.cgt.7700284 PMID: 11332987
79. Mansour M, Palese P and Zamarin D (2011) Oncolytic specificity of newcastle disease virus is mediated by selectivity for apoptosis-resistant cells J Virol 85(12) 6015–6023 https://doi.org/10.1128/JVI.01537-10 PMID: 21471241 PMCID: 3126310
80. Farassati F, Yang AD and Lee PWK (2001) Oncogenes in Ras signalling pathway dictate host-cell permissiveness to herpes simplex virus 1 Nat Cell Biol 3(8) 745–750 https://doi.org/10.1038/35087061 PMID: 11483960
81. DeWeese TL, van der Poel H, and Li S, et al (2001) A phase I trial of CV706, a replication-competent, PSA selective oncolytic adenovirus, for the treatment of locally recurrent prostate cancer following radiation therapy Cancer Res 61(20) 7464–7472 PMID: 11606381
82. Baertsch MA, Leber MF, and Bossow S, et al (2014) MicroRNA-mediated multi-tissue detargeting of oncolytic measles virus Cancer Gene Ther 21(9) 373–380 https://doi.org/10.1038/cgt.2014.40 PMID: 25145311
83. Yu X, Odenthal M and Fries JWU (2016) Exosomes as miRNA carriers: formation-function-future Int J Mol Sci 17(12) 2028 https://doi.org/10.3390/ijms17122028
84. Cimino-Mathews A, Thompson E, and Taube JM, et al (2016) PD-L1 (B7-H1) expression and the immune tumor microenvironment in primary and metastatic breast carcinomas Hum Pathol 47(1) 52–63 https://doi.org/10.1016/j.humpath.2015.09.003 PMCID: 4778421
85. Voorwerk L, Slagter M, and Horlings HM, et al (2019) Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: the TONIC trial Nat Med 25(6) 920–928 https://doi.org/10.1038/s41591-019-0432-4 PMID: 31086347
86. Shen Y and Nemunaitis J (2006) Herpes simplex virus 1 (HSV-1) for cancer treatment Cancer Gene Ther 13(11) 975–992 https://doi.org/10.1038/sj.cgt.7700946 PMID: 16604059
87. Hu JC, Coffin RS, and Davis CJ, et al (2006) A phase I study of OncoVEXGM-CSF, a second-generation oncolytic herpes simplex virus expressing granulocyte macrophage colony-stimulating factor Clin Cancer Res 12(22) 6737–6747 https://doi.org/10.1158/1078-0432.CCR-06-0759 PMID: 17121894
88. Ghouse SM, Nguyen H-M, and Bommareddy PK, et al (2020) Oncolytic herpes simplex virus encoding IL12 controls triple-negative breast cancer growth and metastasis Front Oncol 10 384 https://doi.org/10.3389/fonc.2020.00384 PMID: 32266155 PMCID: 7105799
89. Teshigahara O, Goshima F, and Takao K, et al (2004) Oncolytic viral therapy for breast cancer with herpes simplex virus type 1 mutant HF 10 J Surg Oncol 85(1) 42–47 https://doi.org/10.1002/jso.20005
90. Tan G, Kasuya H, and Sahin TT, et al (2015) Combination therapy of oncolytic herpes simplex virus HF10 and bevacizumab against experimental model of human breast carcinoma xenograft Int J Cancer 136(7) 1718–1730 https://doi.org/10.1002/ijc.29163
91. Kimata H, Imai T, and Kikumori T, et al (2006) Pilot study of oncolytic viral therapy using mutant herpes simplex virus (HF10) against recurrent metastatic breast cancer Ann Surg Oncol https://doi.org/10.1245/ASO.2006.08.035 PMID: 16865590
92. Nemunaitis J, Senzer N, and Sarmiento S, et al (2007) A phase I trial of intravenous infusion of ONYX-015 and enbrel in solid tumor patients Cancer Gene Ther 14(11) 885–893 https://doi.org/10.1038/sj.cgt.7701080 PMID: 17704755
93. Yan Y, Xu H, and Wang J, et al (2019) Inhibition of breast cancer cells by targeting E2F-1 gene and expressing IL15 oncolytic adenovirus Biosci Rep 39(7) https://doi.org/10.1042/BSR20190384
94. Ang L, Guo L, and Wang J, et al (2020) Oncolytic virotherapy armed with an engineered interfering lncRNA exhibits antitumor activity by blocking the epithelial mesenchymal transition in triple-negative breast cancer Cancer Lett 479 42–53 https://doi.org/10.1016/j.canlet.2020.03.012 PMID: 32200038
95. Li Y, Xiao F, and Zhang A, et al (220) Oncolytic adenovirus targeting TGF-β enhances anti-tumor responses of mesothelin-targeted chimeric antigen receptor T cell therapy against breast cancer Cell Immunol 348 104041 https://doi.org/10.1016/j.cellimm.2020.104041
96. Gollamudi R, Ghalib MH, and Desai KK, et al (2010) Intravenous administration of Reolysin, a live replication competent RNA virus is safe in patients with advanced solid tumors Invest New Drugs 28(5) https://doi.org/10.1007/s10637-009-9279-8
97. Bernstein V, Ellard SL, and Dent SF, et al (2018) A randomized phase II study of weekly paclitaxel with or without pelareorep in patients with metastatic breast cancer: final analysis of Canadian Cancer Trials Group IND.213 Breast Cancer Res Treat 167(2) 485–493https://doi.org/10.1007/s10549-017-4538-4
98. Mostafa AA, Meyers DE, and Thirukkumaran CM, et al (2018) Oncolytic reovirus and immune checkpoint inhibition as a novel immunotherapeutic strategy for breast cancer Cancers (Basel) 10(6) 205 https://doi.org/10.3390/cancers10060205
99. Scholl SM, Balloul JM, and Le Goc G, et al (2000) Recombinant vaccinia virus encoding human MUC1 and IL2 as immunotherapy in patients with breast cancer J Immunother https://doi.org/10.1097/00002371-200009000-00007 PMID: 11001550
100. Wang G, Kang X, and Dai S, et al (2020) An engineered oncolytic virus expressing PD-L1 inhibitors activates tumor neoantigen-specific T cell responses Nat Commun 11(1) 1395 https://doi.org/10.1038/s41467-020-15229-5 PMID: 32170083 PMCID: 7070065
101. Zeh HJ, Downs-Canner S, and McCart JA, et al (2015) First-in-man study of western reserve strain oncolytic vaccinia virus: Safety, systemic spread, and antitumor activity Mol Ther 23(1) 202–214 https://doi.org/10.1038/mt.2014.194 PMCID: 4426804
102. Mohamed Amin Z, Ani MAC, and Tan SW, et al (2019) Evaluation of a recombinant newcastle disease virus expressing human IL12 against human breast cancer Sci Rep 9 https://doi.org/10.1038/s41598-019-50222-z PMID: 31570732 PMCID: 6768883
103. Al-Shammari AM, Abdullah AH, and Allami ZM, et al (2019) 2-deoxyglucose and newcastle disease virus synergize to kill breast cancer cells by inhibition of glycolysis pathway through glyceraldehyde3-phosphate downregulation Front Mol Biosci 6 90 https://doi.org/10.3389/fmolb.2019.00090 PMID: 31612140 PMCID: 6777003
104. Zhou C, Tan L, and Sun Y, et al (2019) Exosomes carry microRNAs into neighboring cells to promote diffusive infection of newcastle disease virus Viruses 11(6) 527 https://doi.org/10.3390/v11060527 PMCID: 6631457
105. Ganar K, Das M, and Sinha S, et al (2014) Newcastle disease virus: current status and our understanding Virus Res 184 71–81 https://doi.org/10.1016/j.virusres.2014.02.016 PMID: 24589707 PMCID: 7127793
106. Laurie SA, Bell JC, and Atkins HL, et al (2006) A phase 1 clinical study of intravenous administration of PV701, an oncolytic virus, using two-step desensitization Clin Cancer Res 12(8) 2555–2562 https://doi.org/10.1158/1078-0432.CCR-05-2038 PMID: 16638865
107. Skelding KA, Barry RD, and Shafren DR (2009) Systemic targeting of metastatic human breast tumor xenografts by Coxsackievirus A21 Breast Cancer Res Treat 113(1) 21–30 https://doi.org/10.1007/s10549-008-9899-2
108. Sagara M, TakishimaY, and Miyamoto S, et al (2016) Novel recombinant coxsackievirus B3 infection elicits robust oncolytic activity against human non-small lung cancer and triple-negative breast cancer Mol Ther [http://resolver.ebscohost.com/openurl?sid=EMBASE&issn=15250016&id=doi:10.1038/mt.2016.78&atitle=Novel recombinant coxsackievirus B3 infection elicits robust oncolytic activity against human non-small lung cancer and triple-negative breast cancer&stitle=Mol. Ther.&title=Molecular Therapy&volume=24&issue=&spage=S162&epage=&aulast=Sagara&aufirst=Miyako&auinit=M.&aufull=Sagara M.&coden=&isbn=&pages=S162-&date=2016&auinit1=M&auinitm=] https://doi.org/10.1016/S1525-0016(16)33218-X
109. Holl EK, Brown MC, and Boczkowski D, et al (2016) Recombinant oncolytic poliovirus, PVSRIPO, has potent cytotoxic and innate inflammatory effects, mediating therapy in human breast and prostate cancer xenograft models Oncotarget 7(48) 79828–79841c https://doi.org/10.18632/oncotarget.12975 PMID: 27806313 PMCID: 5346754
110. Dong X, Qu W, and Ma S, et al (2013) Potent antitumoral effects of targeted promoter-driven oncolytic adenovirus armed with Dm-dNK for breast cancer in vitro and in vivo Cancer Lett 328(1) 95–103 https://doi.org/10.1016/j.canlet.2012.09.003
111. Iankov ID, Kurokawa CB, and D’Assoro AB, et al (2015) Inhibition of the Aurora A kinase augments the anti-tumor efficacy of oncolytic measles virotherapy Cancer Gene Ther 22(9) 438–444 https://doi.org/10.1038/cgt.2015.36 PMID: 26272026 PMCID: 4589445
112. Iankov ID, Msaouel P, and Allen C, et al (2010) Demonstration of anti-tumor activity of oncolytic measles virus strains in a malignant pleural effusion breast cancer model Breast Cancer Res Treat 122(3) 745–754 https://doi.org/10.1007/s10549-009-0602-z PMCID: 2935656
113. Brun J, McManus D, and Lefebvre, et al (2010) Identification of genetically modified maraba virus as an oncolytic rhabdovirus Mol Ther 18(8) 1440–1449 https://doi.org/10.1038/mt.2010.103 PMID: 20551913 PMCID: 2927055