Adaptive immunity in cancer immunology and therapeutics
Emma L Spurrell1 and Michelle Lockley2
1Whittington Health NHS Trust, Magdala Avenue, London N19 5NF, UK
2Barts Cancer Institute, Queen Mary University of London Charterhouse Square, London EC1M 6BQ, UK
Correspondence to: Emma Spurrell. Email: emmaspurrell@doctors.org.uk
Abstract
The vast genetic alterations characteristic of tumours produce a number of tumour antigens that enable the immune system to differentiate tumour cells from normal cells. Counter to this, tumour cells have developed mechanisms by which to evade host immunity in their constant quest for growth and survival. Tumour-associated antigens (TAAs) are one of the fundamental triggers of the immune response. They are important because they activate, via major histocompatibility complex (MHC), the T cell response, an important line of defense against tumourigenesis. However, the persistence of tumours despite host immunity implies that tumour cells develop immune avoidance. An example of this is the up-regulation of inhibitory immune checkpoint proteins, by tumours, which induces a form of self-tolerance. The majority of monoclonal antibodies in clinical practice have been developed to target tumour-specific antigens. More recently there has been research in the down-regulation of immune checkpoint proteins as a way of increasing anti-tumour immunity.
Keywords: adaptive immunity, tumour-associated antigens, CTLA4, PD-1, PDL-1, monoclonal antibody, immune tolerance
Copyright: © the authors; licensee ecancermedicalscience. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Introduction
Tumours do not grow in isolation, but exist within a complex network of structures, cells, and chemical signals ranging from epithelial cells, stroma, blood, and lymphatic vasculature, immune cells, cytokines, and chemokines. The vast genetic alterations characteristic of tumours produce a number of tumour antigens that enable the immune system to differentiate tumour cells from normal cells. Counter to this, tumour cells have developed mechanisms by which to evade host immunity in their constant quest for growth and survival.
A typical tumour structure includes the tumour core, the invasive margin, and the surrounding stromal and lymphoid components. Within all of these is a heterogeneous immune infiltrate that can be diverse from patient to patient, as well as within different metastatic sites of a single patient. A typical tumour will contain all immune cell-types, including macrophages, dendritic cells, natural killer (NK) cells, mast cells, B cells, and T cells [including T helper 1 (TH1), T helper 2 (TH2), regulatory T cells (TReg) and cytotoxic T cells]. Within this immune milieu there are components that are beneficial, as well as components that are deleterious to the patient. Histopathological analysis of tumours has identified that different immune cells may be found in different locations within a tumour. The variation in density and distribution of immune cells within tumours is thought to affect clinical outcome [1]. Although the innate immune response plays a role in anti-tumour immunity, detailed discussion is beyond the scope of this review, which will focus on the adaptive immune response.
Adaptive immunity and the control of tumour growth
Tumour-associated antigens (TAAs) are one of the fundamental triggers of the immune response. They are important because they activate, via, major histocompatibility complex (MHC), the T cell response, an important line of defense against tumourigenesis.
TAAs that are recognised by T cells are classified in Table 1. The review is restricted to TAAs recognised by T cells as these represent the main therapeutic targets in oncology. Although TAAs arise by different mechanisms, they are all presented to T cells via MHC class I or II on antigen presenting cells. This triggers T cell activation with expression of co-stimulatory molecules and secretion of chemokines and cytokines. The effect is to drive clonal expansion of the T cell as well as to recruit other immune effector cells (including components of the innate immune system). CD4 T cells, also known as T helper cells, secrete cytokines and chemokines that regulate different aspects of the immune response. TH1 CD4 T cells activate CD8 T cells, favouring cellular immunity. TH2 CD4 T cells act on B cells, favouring humoral immunity. CD8 T cells, which are directly cytotoxic, are activated both by direct presentation of antigen, via MHC class I, or via CD4 T cell-mediated activation. Ultimately, the tumour cell is destroyed by direct cell-mediated cytotoxicity as well an indirect antibody complement-mediated cytotoxicity [2].
Immune editing and evasion
The persistence of tumours despite host immunity implies that tumour cells develop immune avoidance. There are a number of mechanisms by which this may occur. Some tumours have been demonstrated to lose expression of MHC molecules making them unable to present tumour antigens, thus evading T cell recognition. Some tumours secrete immunosuppressive cytokines, e.g., IL-10. There are tumours that grow within their own immune-privileged site by generating physical barriers, e.g., collagen and fibrin, thus making them invisible to the immune system.
Tumours can also evade the immune response by up-regulating inhibitory molecules and inducing a form of self-tolerance [4]. Immune checkpoints are vital for maintenance of self-tolerance and protection of normal tissue from damage at the site of an immune response. Specific regulatory cells and inhibitory receptors achieve immune tolerance. Regulatory T cells (TReg) are immunosuppressive. They secrete inhibitory cytokines, such as IL-10 and TGFβ, resulting in down-regulation of effector B and T cells [5]. T cells rely on co-stimulatory signals to generate an immune response. Inhibition of co-stimulatory signals helps to maintain immune tolerance. Cytotoxic T-lymphocyte associated antigen 4 (CTLA4) and programmed cell death protein 1(PD1) are both inhibitory receptors involved in down-regulation of immune responses.
There is currently a lot of interest in the down-regulation of immune checkpoint proteins as a way of increasing anti-tumour immunity. Antibodies against CTLA-4 and PD-1 have been tested in therapeutic trials, which are discussed below.
Table 1. Classification of tumour-associated antigens that are recognised by T cells [3].
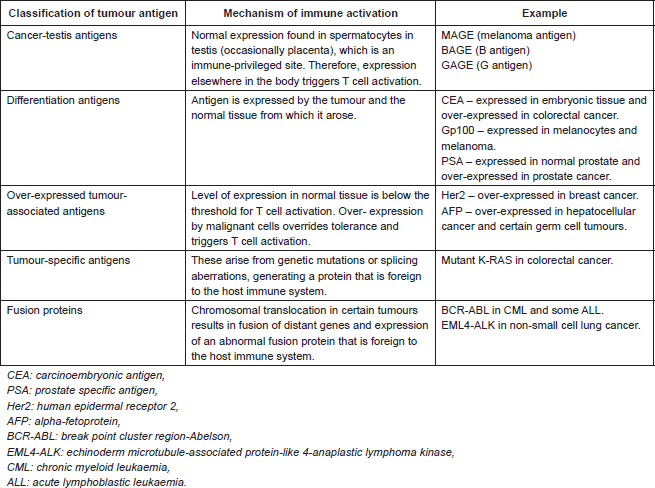
Tumour immunology and clinical outcome
In many cancers there is a demonstrable correlation between level of immune cell infiltration and prognosis. Different populations of cells within the immune infiltrate affect prognosis in different ways. There are a lot of published data correlating prognosis with type of immune cell infiltrate. Table 2 summarises some of the data.
Most recently it has been demonstrated that the higher the proportion of tumour infiltrating lymphocytes on core biopsy of breast cancer, the greater the likelihood of a pathological complete remission following neoadjuvant chemotherapy and Herceptin, in Her2-positive breast cancer. This data was reported at the 2013 San Antonio Breast Cancer Symposium where the team demonstrated a correlation between level of tumour-infiltrating lymphocytes and immune activation as well as clinical outcome, implying a pre-existing anti-tumour immunity. They also reported an association between high expression of inhibitory receptors PD-1 and CTLA-4, and benefit from Herceptin; hypothesizing that Herceptin overcomes tumour-mediated immunosuppression [24]. They used a mouse model of Her2-positive breast cancer to demonstrate improved efficacy of Herceptin when combined with a T-cell checkpoint inhibitor, compared to Herceptin alone [24].
Table 2. Correlation of immune cell infiltrate and clinical outcome.
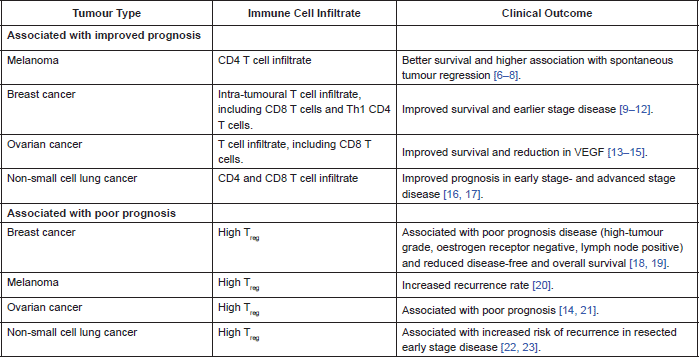
The positive prognostic association of high CD4 and CD8 T cell infiltration within a tumour implies a clinically relevant anti-tumour immune response. This response is subject to down-regulation by inhibitory immune cells, including TReg. Therefore, tumours with high TReg infiltration may experience less of an anti-tumour immune response, hence the association with poorer clinical outcomes.
Targeting tumour antigens with antibodies
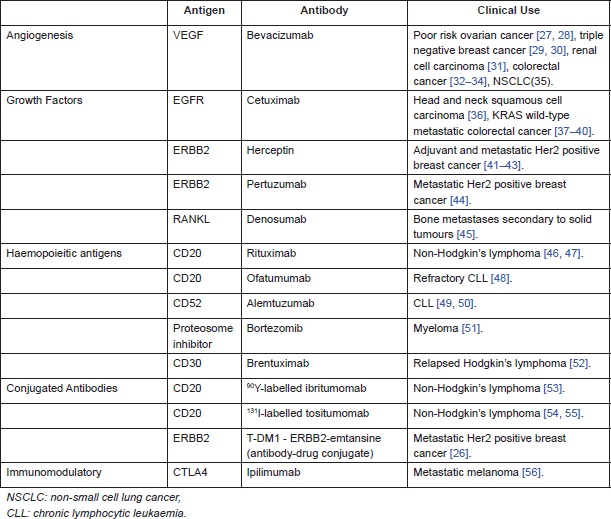
Targeting tumour antigens with antibodies is an established therapeutic for both solid tumours and haematological malignancies. There are different categories of tumour antigens that have been targeted by monoclonal antibodies. In haematological malignancies, antibodies have been raised against cluster of differentiation (CD) markers on T and B cells. In solid tumours, targets include growth factors, e.g., epidermal growth factor receptor (EGFR) and receptor activator of nuclear factor-κB ligand (RANKL), as well as angiogenesis, e.g., vascular endothelial growth factor (VEGF).
There are several mechanisms by which antibodies cause tumour cell death. There is the direct action of the antibody, where binding of antibody to the cell causes receptor blockade. This inhibits the downstream signaling pathways within the cell, ultimately leading to apoptosis. Immune-mediated mechanisms include antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC), where the Fc component (rather than the antigen-binding domain) of the antibody is most important. There is also the immune modulation of T cell function via inhibition of immune checkpoint proteins by targeted antibodies, e.g., anti-CTLA4 and anti-PD1.
Using Herceptin as an example, once the antibody has bound to Her2, expressed on the surface of a breast cancer cell, it prevents the dimerization of the receptor. Receptor dimerization is necessary for the activation of downstream signaling pathways. Herceptin inhibits the MAP kinase and PI3 kinase signaling pathways as well as causing cell cycle arrest, ultimately leading to apoptosis. The Fc component of Herceptin can trigger the innate immune response, in particular, NK cells which are directly cytotoxic to the tumour cell. Internalisation of Herceptin into the cell results in degradation of the antibody and presentation of antibody proteins to T cells via MHC. T cells are not only directly cytotoxic but will recruit other immune cells, including B cells, leading to ADCC. Finally, the Herceptin–Her2 complex results in activation of the complement pathway with binding of C1q to the antibody–antigen complex, resulting in cell lysis [25].
There are antibodies that have been conjugated to a radioisotope or cytotoxic agent to deliver these directly to the tumour cell in therapeutic doses. Conjugation of a radioisotope to antibody is used as a therapeutic in lymphoma. Most recently, T-DM1 (trastuzumab-emtansine) has shown clinical benefit in Her2-positive metastatic breast cancer where trastuzumab is conjugated to an anti-microtubule agent, emtansine. Upon ligand binding, the antibody conjugate becomes internalised into the cell enabling targeted delivery of a cytotoxin. This achieves delivery of a cytotoxic dose of a therapeutic agent directly to its target, whilst sparing healthy tissue, hence minimising toxicity [26].
Table 3 summarises examples of antibodies used in clinical practice, either NICE (National Institute for Health and Care Excellence) approved or available via the Cancer Drugs Fund (government funding of drugs not yet approved by NICE, available in England).
Table 3. Monoclonal antibodies used in clinical practice.
Targeting immune checkpoint proteins with antibodies
Anti- CTLA4
Ipilimumab is a monoclonal antibody that blocks CTLA4 with the aim of promoting anti-tumour immunity. CTLA4 is expressed on T cells, where it serves to regulate the magnitude of the T cell response. Once the T cell receptor has bound target antigen, the co-stimulatory receptor CD28 amplifies T cell signaling. Activated T cells are not only directly cytotoxic but also act to recruit other components of both the innate and adaptive immune response.CTLA4 counteracts the activity of the CD28 receptor thus down-regulating individual T cells and preventing recruitment of other T and immune effector cells. TReg highly express CTLA4, which enhances their proliferation and immunosuppressive activity [57].
Ipilimumab has shown the greatest clinical activity in metastatic melanoma. The pivotal phase III trial demonstrating a survival benefit with ipilimumab was published in 2010. There were three arms in the trial, ipilimumab alone, ipilimumab with gp100 (melanoma cancer vaccine), and gp100 alone. The two arms treated with ipilimumab showed the same survival suggesting that the active agent was the ipilumumab. The patients treated with ipilimumab had a significant survival advantage to those treated with gp100 alone (10.1 months vs. 6.4 months, p = 0.003) [56]. Subsequent analyses have shown the survival benefits to be durable. A pooled analysis of 1,861 patients with melanoma treated with ipilimumab in 12 prospective and retrospective studies showed that 22% were still alive at 3 years, 17% were alive at 7 years, and the longest recorded survival in the database was 9.9 years [58].
The majority of toxicities experienced with ipilimumab were immune-related, mostly affecting skin and GI tract. Diarrhoea was the most common immune-related toxicity, occurring in 31% of patients. This manifested as frank colitis in some patients, requiring corticosteroids or infliximab (anti-TNF). Other immune-related events included skin rash, vitiligo, and endocrine insufficiencies (thyroid, pituitary, and adrenal) [56].
Anti-PD-1
PD-1 is expressed on T cells, particularly TReg, its expression being induced when T cells become activated. PD-1 primarily functions in peripheral tissues where T cells are exposed to the immunosuppressive PD-1 ligands, PD-L1 and PD-L2, that are expressed by tumour cells and surrounding stroma. Once PD-1 has bound one of its ligands, it functions to inhibit kinases responsible for T cell activation, except in TReg where binding of PD-1 to ligand enhances their proliferation [57, 59].
Blockade of PD-1 by anti-PD-1 antibody has been tested in phase I. The largest cohort tested included 296 patients with advanced solid tumours, including melanoma, NSCLC, renal cell carcinoma, prostate cancer, and colorectal cancer. Responses were observed in 20–25% of patients with NSCLC, renal call carcinoma, and melanoma. No responses were observed in patients whose tumour specimens were negative for PD-L1. Similar to ipilimumab, responses were durable in some patients, with response duration of a year or more [59]. This is impressive given the fact that all patients were heavily pre-treated and no longer responding to conventional therapy.
The most common treatment-related toxicities seen with anti-PD-1 were fatigue, reduced appetite, diarrhoea, nausea, rash, and pruritus. Grade 3 or 4 treatment-related adverse events occurred in 14% of patients. Like ipilimumab, anti-PD-1 was associated with immune-related toxicity including pneumonitis, vitiligo, colitis, hepatitis, hypophysitis, and thyroiditis. There were three deaths from pulmonary toxicity [59].
There is ongoing interest in targeting PD-1 in different ways, for example, targeting one of the ligands with an anti-PD-L1 antibody, as well as combining blockade of PD-1 and CTLA4 [60].
Conclusion
The tumour microenvironment consists of a complex milieu of cells, including immune cells. Some components of the immune infiltrate exert a beneficial anti-tumour effect, while others can down-regulate host immunity, and promote tumour-immune evasion. The proportion of the different infiltrates within a tumour has been shown to affect clinical outcome.
Antibody therapy has successfully targeted tumour antigens for many years. Targeted antibodies exert their therapeutic effect not only by inhibiting the target but also by activation of the host immune system.There is now increasing interest in using antibodies to up-regulate host anti-tumour immunity with some durable results seen in early trials so far.
References
1. Fridman WH et al (Apr 2012) The immune contexture in human tumours: impact on clinical outcome Nat Rev Cancer 12(4): 298–306 PMID: 22419253
2. Janeway C (2005) Immunobiology: the immune system in health and disease 6th edn (Churchill Livingstone)
3. Novellino L, Castelli C and Parmiani G (March 2005) A listing of human tumor antigens recognized by T cells: March 2004 update Cancer Immunol Immunother 54(3):187–207 PMID: 15309328
4. Zou W, Chen L (Jun 2008) Inhibitory B7-family molecules in the tumour microenvironment Nat Rev Immunol 8(6):467–77 PMID: 18500231
5. Vignali DA, Collison LW and Workman CJ (July 2008) How regulatory T cells work Nat Rev Immunol 8(7):523-32 PMID: 18566595 PMCID: 2665249
6. Clemente CG et al (April 1 1996) Prognostic value of tumor infiltrating lymphocytes in the vertical growth phase of primary cutaneous melanoma Cancer 77(7) 1303–10 PMID: 8608507
7. Hillen F et al (Jan 2008) Leukocyte infiltration and tumor cell plasticity are parameters of aggressiveness in primary cutaneous melanoma Cancer Immunol Immunother 57(1):97–106 PMID: 17602225
8. Tefany FJ et al (Aug 1991) Immunocytochemical analysis of the cellular infiltrate in primary regressing and non-regressing malignant melanoma J Invest Dermatol 97(2) 197–202 PMID: 1712819
9. Alexe G et al (Nov 12 2007) High expression of lymphocyte-associated genes in node-negative HER2 breast cancers correlates with lower recurrence rates Cancer Res 67(22) 10669–76 PMID: 18006808
10. Mahmoud SM et al (May 2011) Tumor-infiltrating CD8 lymphocytes predict clinical outcome in breast cancer J Clin Oncol 29(15):1949–55 PMID: 21483002
11. Marrogi AJ et al (Oct 21 1997) Study of tumor infiltrating lymphocytes and transforming growth factor-beta as prognostic factors in breast carcinoma Int J Cancer 74(5) 492–501 PMID: 9355970
12. Oldford SA et al (Nov 2006) Tumor cell expression of HLA-DM associates with a Th1 profile and predicts improved survival in breast carcinoma patients Int Immunol 18(11) 1591–602 PMID: 16987935
13. Kusuda T et al (Jun 2005) Relative expression levels of Th1 and Th2 cytokine mRNA are independent prognostic factors in patients with ovarian cancer. Oncol Rep 13(6) 1153–8 PMID: 15870936
14. Sato E et al (Dec 20, 2005) Intraepithelial CD8 tumor-infiltrating lymphocytes and a high CD8 /regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci USA 102(51) 18538–43 PMID: 16344461 PMCID: 1311741
15. Zhang L et al (Jan 16 2003) Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer N Engl J Med 348(3) 203–13 PMID: 12529460
16. Al-Shibli KI et al (Aug 15 2008) Prognostic effect of epithelial and stromal lymphocyte infiltration in non-small cell lung cancer Clin Cancer Res 14(16) 5220–7 PMID: 18698040
17. Kawai O et al (Sep 15, 2008) Predominant infiltration of macrophages and CD8( ) T Cells in cancer nests is a significant predictor of survival in stage IV nonsmall cell lung cancer Cancer 113(6) 1387–95 PMID: 18671239
18. Bates GJ et al (Dec 1 2006) Quantification of regulatory T cells enables the identification of high-risk breast cancer patients and those at risk of late relapse J Clin Oncol 24(34) 5373–80 PMID: 17135638
19. Gobert M et al (Mar 1 2009) Regulatory T cells recruited through CCL22/CCR4 are selectively activated in lymphoid infiltrates surrounding primary breast tumors and lead to an adverse clinical outcome Cancer Res 69(5) 2000–9 PMID: 19244125
20. Miracco C et al (Nov 2007) Utility of tumour-infiltrating CD25 FOXP3 regulatory T cell evaluation in predicting local recurrence in vertical growth phase cutaneous melanoma Oncol Rep 18(5) 1115–22 PMID: 17914561
21. Hamanishi J et al (Feb 27, 2007) Programmed cell death 1 ligand 1 and tumor-infiltrating CD8 T lymphocytes are prognostic factors of human ovarian cancer Proc Natl Acad Sci USA 104(9)3360–5 PMID: 17360651 PMCID: 1805580
22. Petersen RP et al (Dec 15 2006) Tumor infiltrating Foxp3 regulatory T-cells are associated with recurrence in pathologic stage I NSCLC patients Cancer 107(12):2866-72 PMID: 17099880
23. Shimizu K et al (May 2010) Tumor-infiltrating Foxp3 regulatory T cells are correlated with cyclooxygenase-2 expression and are associated with recurrence in resected non-small cell lung cancer J Thorac Oncol 5(5) 585–90 PMID: 20234320
24. Loi S MS and Salgado R et al (2013) Tumor infiltrating lymphocytes (TILs) indicate trastuzumab benefit in early-stage HER2-positive breast cancer (HER2 BC) San Antonio Breast Cancer Symposium; San Antonio, USA.
25. Nuti M and Bellati F et al (2011) Immune effects of trastuzumab J Cancer 2 317–23 DOI: 10.7150/jca.2.317 PMID: 21716848 PMCID: 3119394
26. Verma S et al (Nov 8 2012) Trastuzumab emtansine for HER2-positive advanced breast cancer N Engl J Med 367(19) 1783–91 PMID: 23020162
27. Burger RA et al (Dec 29, 2011) Incorporation of bevacizumab in the primary treatment of ovarian cancer N Engl J Med 365(26) 2473–83 PMID: 22204724
28. Perren TJ et al (Dec 29 2011) A phase 3 trial of bevacizumab in ovarian cancer N Engl J Med 365(26) 2484–96. PMID: 22204725
29. Miles DW et al (Jul 10 2010) Phase III study of bevacizumab plus docetaxel compared with placebo plus docetaxel for the first-line treatment of human epidermal growth factor receptor 2-negative metastatic breast cancer J Clin Oncol 28(20) 3239–47 PMID: 20498403
30. Miller K et al (Dec 27, 2007) Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer N Engl J Med 357(26) 2666–76 PMID: 18160686
31. Escudier B et al (May 1 2010) Phase III trial of bevacizumab plus interferon alfa-2a in patients with metastatic renal cell carcinoma (AVOREN): final analysis of overall survival J Clin Oncol 28(13) 2144–50 PMID: 20368553
32. Giantonio BJ et al (Apr 20 2007) Bevacizumab in combination with oxaliplatin, fluorouracil, and leucovorin (FOLFOX4) for previously treated metastatic colorectal cancer: results from the Eastern Cooperative Oncology Group Study E3200 J Clin Oncol 25(12)1539–44 PMID: 17442997
33. Hurwitz H et al (Jun 3 2004) Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer N Engl J Med 350(23) 2335–42 PMID: 15175435
34. Saltz LB et al (Apr 20, 2008) Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study J Clin Oncol 26(12) 2013–9 PMID: 18421054
35. Reck M et al (Sep 2010) Overall survival with cisplatin-gemcitabine and bevacizumab or placebo as first-line therapy for nonsquamous non-small-cell lung cancer: results from a randomised phase III trial (AVAiL) Ann Oncol 21(9) 1804–9 PMID: 20150572 PMCID: 2924992
36. Bonner JA et al (Feb 9 2006) Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck N Engl J Med 354(6) 567–78 PMID: 16467544
37. Bokemeyer C et al (Feb 10 2009) Fluorouracil, leucovorin, and oxaliplatin with and without cetuximab in the first-line treatment of metastatic colorectal cancer J Clin Oncol 27(5) 663–71 PMID: 19114683
38. Cunningham D et al (Jul 22 2004) Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer N Engl J Med 351(4) 337–45 PMID: 15269313
39. Karapetis C et al (Oct 23 2008) K-ras mutations and benefit from cetuximab in advanced colorectal cancer N Engl J Med 359(17) 1757–65 PMID: 18946061
40. Maughan TS et al (Jun 18 2011) Addition of cetuximab to oxaliplatin-based first-line combination chemotherapy for treatment of advanced colorectal cancer: results of the randomised phase 3 MRC COIN trial Lancet 2011 377(9783) 2103–14 PMID: 21641636 PMCID: 3159415
41. Gianni L et al (Mar 12 2011) Treatment with trastuzumab for 1 year after adjuvant chemotherapy in patients with HER2-positive early breast cancer: a 4-year follow-up of a randomised controlled tria. Lancet Oncol 12(3) 236–44 PMID: 21354370
42. Slamon D et al (Oct 6 2011) Adjuvant trastuzumab in HER2-positive breast cancer N Engl J Med 365(14):1273–83 PMID: 21991949 PMCID: 3268553
43. Slamon DJ et al (Mar 15 2001) Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2 N Engl J Med 344(11)783–92 PMID: 11248153
44. Baselga J et al (Jan 12 2012) Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer N Engl J Med 366(2)109–19 PMID: 22149875
45. Henry D et al (Oct 26 2013) Delaying skeletal-related events in a randomized phase 3 study of denosumab versus zoledronic acid in patients with advanced cancer: an analysis of data from patients with solid tumors Support Care Cancer PMID: 24162260
46. Coiffier B et al (Jan 24 2002) CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma N Engl J Med 346(4) 235–42 PMID: 11807147
47. McLaughlin P et al (Aug 1998) Rituximab chimeric anti-CD20 monoclonal antibody therapy for relapsed indolent lymphoma: half of patients respond to a four-dose treatment program J Clin Oncol 16(8) 2825–33 PMID: 9704735
48. Wierda WG et al (Nov 10 2011) Ofatumumab is active in patients with fludarabine-refractory CLL irrespective of prior rituximab: results from the phase 2 international study Blood 118(19) 5126–9 PMID: 21856867
49. Hillmen P et al (Dec 10 2007) Alemtuzumab compared with chlorambucil as first-line therapy for chronic lymphocytic leukemia J Clin Oncol 25(35) 5616–23 PMID: 17984186
50. Keating MJ et al (May 15 2002) Therapeutic role of alemtuzumab (Campath-1H) in patients who have failed fludarabine: results of a large international study Blood 99(10) 3554–61 PMID: 11986207
51. Oakervee HE et al (Jun 2005) PAD combination therapy (PS-341/bortezomib, doxorubicin and dexamethasone) for previously untreated patients with multiple myeloma Br J Haematol 129(6) 755-62 PMID: 15953001
52. Younes A et al Results of a pivotal phase II study of brentuximab vedotin for patients with relapsed or refractory Hodgkin's lymphoma J Clin Oncol 30(18) 2183–9 PMID: 22454421 PMCID: 3646316
53. Gordon LI, Molina A et al (Jun 15 2004) Durable responses after ibritumomab tiuxetan radioimmunotherapy for CD20 B-cell lymphoma: long-term follow-up of a phase 1/2 study Blood 103(12) 4429–31 PMID: 15016644
54. Kaminski MS et al (Oct 1 2001) Pivotal study of iodine I 131 tositumomab for chemotherapy-refractory low-grade or transformed low-grade B-cell non-Hodgkin's lymphomas J Clin Oncol 19(19) 3918–28 PMID: 11579112
55. Press OW et al (Sep 1 2006) Phase II trial of CHOP chemotherapy followed by tositumomab/iodine I-131 tositumomab for previously untreated follicular non-Hodgkin's lymphoma: five-year follow-up of Southwest Oncology Group Protocol S9911 J Clin Oncol 24(25) 4143–9 PMID: 16896003
56. Hodi FS et al (Aug 19 2010) Improved survival with ipilimumab in patients with metastatic melanoma N Engl J Med 363(8) 711–23 PMID: 20525992 PMCID: 3549297
57. Pardoll DM (Apr 2010) The blockade of immune checkpoints in cancer immunotherapy Nat Rev Cancer 12(4) 252–64 PMID: 22437870
58. From ECC2013 (Dec 2013) Skin cancer: Ipilimumab-treated patients survive up to 10 years Nat Rev Clin Oncol 10(12) 669
59. Topalian SL et al (Jun 28 2012) Safety, activity, and immune correlates of anti-PD-1 antibody in cancer N Engl J Med 366(26) 2443–54 PMID: 22658127 PMCID: 3544539
60. Wolchok JD et al (Jul 11 2013) Nivolumab plus ipilimumab in advanced melanoma N Engl J Med 369(2)122–33 PMID: 23724867

