Thymoma associated with hypogammaglobulinaemia and pure red cell aplasia
Juan Briones1, Mirentxu Iruretagoyena2, Héctor Galindo1, Claudia Ortega3, Pablo Zoroquiain4, José Valbuena4, Francisco Acevedo1, Mauricio Ocqueteau1 and Cesar Sánchez1
1 Department of Hematology-Oncology, School of Medicine, Pontifical Catholic University of Chile, Chile 8330024
2 Department of Clinical and Rheumatologic Immunology, School of Medicine, Pontifical Catholic University of Chile, Chile 8330024
3 Department of Radiology, School of Medicine, Pontifical Catholic University of Chile, Chile 8330024
4 Department of Pathological Anatomy, School of Medicine, Pontifical Catholic University of Chile, Chile 8330024
Correspondence to: Dr Cesar Sánchez. E-mail: csanchez@med.puc.cl
Abstract
Thymomas are neoplasias that begin in the thymus and develop in the anterior mediastinum. They are commonly associated with a variety of systemic and autoimmune disorders, such as pure red cell aplasia, hypogammaglobulinaemia, pancytopaenia, collagen diseases, and, most commonly, myasthenia gravis. The presence of inter-current infections, especially diarrhoea and pneumonia, in the presence of lymphocyte B depletion and hypogammaglobulinaemia is known as Good’s syndrome and may affect up to 5% of patients with thymoma. While anaemia is present in 50%–86% of patients with Good’s syndrome, only 41.9% of cases present pure red cell aplasia. Concomitance of these two conditions has only been rarely studied.
We report on the case of a 55-year-old man diagnosed with advanced thymoma, who, during the progression of his disease, developed signs and symptoms suggesting Good’s syndrome and pure red cell aplasia. We also performed a brief review of the literature concerning this association, its clinical characteristics, and treatment.
Keywords: thymoma, pure red cell aplasia, hypogammaglobulinaemia.
Introduction
Thymomas are not common neoplasias, with an incidence of 0.15 cases per 100,000 people per year. They represent 20%–30% of mediastinal tumours in adults [1, 2]. Patients usually present with symptoms secondary to local compression, such as respiratory symptoms or superior vena cava syndrome. On other occasions, they may appear with a paraneoplastic syndrome [2], including myasthenia gravis, pure red cell aplasia, connective tissue disorders, and hypogammaglobulinaemia/Good’s syndrome [1, 2]. Good’s syndrome affects at least 5% of patients with thymomas [3]. The principal immunological abnormalities described for this syndrome include hypogammaglobulinaemia, B-cell lymphopaenia, CD4/CD8 ratio inversion, low T CD4 lymphocyte counts, and alteration of the mitogenic T cell response [4]. Pure red cell aplasia is described in less than 10% of patients with thymoma and is characterised by an erythropoiesis insufficiency, with granulopoiesis and megakariopoiesis conservation [11]. Pure red cell aplasia may be the initial finding that leads to the diagnosis of thymoma or may develop after diagnosis.
We report the case of a 55-year-old man, diagnosed with advanced thymoma, who developed hypogammaglobulinaemia/Good’s syndrome and pure red cell aplasia as his condition evolved.
Clinical case
The case involved a 53-year-old male patient, with a history of type 2 diabetes mellitus 2 and dyslipidaemia. He consulted his doctor for a month-long condition of dry cough associated with a loss of weight amounting to 10 kg. A chest x-ray showed a rise of the left hemidiaphragm. A computed tomography (CT) scan of the thorax showed an anterior mediastinal tumour, with no evidence of spreading to adjacent tissue (Figure 1). A biopsy of the mediastinal tumour, guided by the CT, was performed, confirming the diagnosis of thymoma, which was treated by thymectomy that left residual macroscopic disease. A biopsy of the surgical piece showed type B1 thymoma, according to the World Health Organization (WHO) classification, with infiltration into the sub-pericardial connective tissue and mediastinal adipose tissue, associated with pulmonary implants (Figure 2AD) Masaoka stage III. Positron emission tomography-computed tomography (PET-CT) control after three months showed no evidence of local or systemic spread of the thymoma, and so it was decided to maintain clinical controls. During the followup period, the patient presented with two episodes of pneumonia.
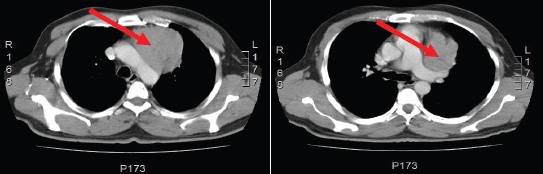
Figure 1: Computed Tomography of the chest shows the presence of an anterior mediastinal solid mass (red arrow) that has a maximum diameter of 7.5 cm. This tumor has a fine line of separation from adjacent vascular structure and back side of the sternum and rib cartilage.
A year after surgery, the patient developed exercise-induced dyspnoea, associated with a progressive deterioration in functional capacity. The chest CT (Figure 3) showed left nodular pleural thickening, associated with pulmonary ipsilateral and adenopathic mediastinal nodules, compatible with a diagnosis of tumour recurrence. Due to symptomatic thymoma progression, palliative cyclophosphamide, doxorubicin, cisplatin, and prednisone combination chemotherapy (CAPPr) was initiated.
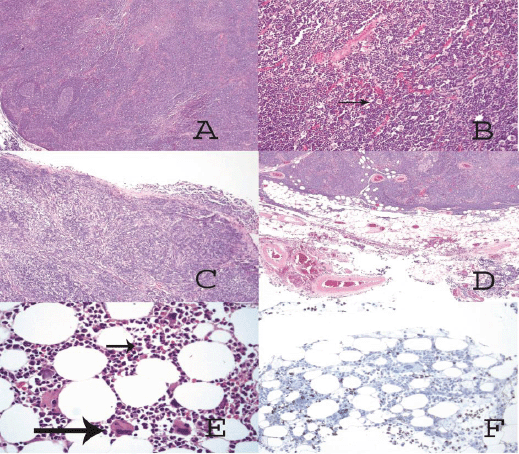
Figure 2: (A) Solid tumor made up of lymphocytes with germinal center formation. (B) At higher magnification, (arrow) large cells of clear eosinophilic cytoplasm and round nuclei (epithelial cells) are also observed. (C) The tumor has invaded the pleura, passing through it. (D) The tumor has invaded the mediastinal adipose tissue. (E) In the hypocellular bone marrow only megakaryocytes (big arrow) and myeloid elements are observed (short arrow). (F) Immunohistochemical staining against glycophorin C is only positive on red cells; no erythroid precursor-like cells are observed.
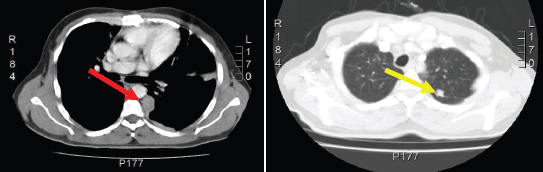
Figure 3: Computed Tomography of the chest shows left pleural nodular thickening predominantly in costophrenic recesses, identifying large tumors at this level (red arrow). Multiple left juxta-cisural nodules up to 13 mm (yellow arrow).
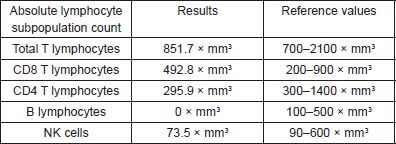
Prior to the beginning of treatment, the patient developed the signs and symptoms of pneumonia and responded favourably to treatment with antibiotics. After the first and second cycles with CAPPr, the patient had to be hospitalised with febrile neutropaenia; both times, it was satisfactorily treated with antibiotics and colony-stimulating factors. After the third cycle with CAPPr, the patient was once again hospitalised, due to symptoms of abdominal pain and abundant diarrhoea (30 episodes per day), containing mucous and blood. A general laboratory analysis did not detect neutropaenia (Table 1). The microbiological study was negative. The study was completed with a colonoscopy that showed an isolated blind ulcer, the biopsy of which showed the presence of lamina propria micro-haemorrhaging. Suspecting acquired hypogammaglobulinaemia, an immunoglobulin count (Table 2) and lymphocyte subpopulation analysis (Table 3) were requested, which confirmed the diagnosis of Good’s syndrome. As a result, the patient was given an intravenous immunoglobulin supplement. He was discharged in good condition to continue outpatient management.
Three months later, the patient was again hospitalised with the signs and symptoms of anaemic syndrome. The complete blood count (CBC) showed severe arregenerative anaemia (Table 1). He was given a transfusion of red blood cells, and a bone marrow biopsy was performed, showing typical findings consistent with pure red cell aplasia. (Figure 2E,F). Evaluation revealed radiological progression of the disease, so chemotherapy with second-line drugs paclitaxel and carboplatin was given. Since his release, he has not returned to the clinic.
Discussion
The relationship between thymoma and hypogammaglobulinaemia was initially described by Dr Robert Good in 1954 [4]. At the present time, there are various definitions for Good’s syndrome. Some authors define it as a subtype of common variable immunodeficiency (CVID); however, the depletion of peripheral B lymphocytes associated with Good’s syndrome is not characteristic of CVID, which is typically associated with an alteration in B lymphocyte maturation [5, 6]. Others define it as hypogammaglobulinaemia associated with thymoma, which is consistent with the case originally described by Dr Good [5].
Table 1. General laboratory analysis.
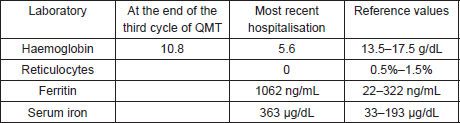
Table 2. Immunoglobulin counts.
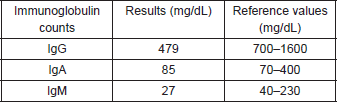
Table 3. Lymphocyte subpopulation.
As previously mentioned, patients with Good’s syndrome have, in the presence of thymoma, B cells missing or diminished in peripheral blood, hypogammaglobulinaemia and cellular immunity defects [2, 7]. Immunodeficiency is combined, affecting the humoral response as well as the cellular, predisposing patients to upper and lower respiratory infections, similar to CVID and also to opportunistic infections observed in HIV-positive patients [2, 8, 9]. Good’s syndrome most often affects patients between 40 and 70 years of age [2], affecting both sexes equally and occurring in less than 5% of patients with thymoma [4]. As to prognosis, 70% of Good’s syndrome patients will be alive after five years compared with almost 100% survival for patients with agammaglobulinaemia related to the X chromosome and with CVID [4, 16 ].
According to a systematic review of the literature, diagnosis of thymoma precedes documentation of hypogammaglobulinaemia, infection, or diarrhoea in 42% of patients. In this study, thymoma was observed after diagnosis of hypogammaglobulinaemia or infection in 19.7% of cases. Around 37.9% of the population was diagnosed simultaneously [8].
The pathogenesis of Good’s syndrome is unknown. However, at least three hypotheses have been put forth: (1) cytokines: the first hypothesis arose from murine models, which demonstrated that cytokines such as limitin, an interferon-type cytokine produced by a stromal cell line in the bone marrow, is capable of influencing the growth and differentiation of B cell precursors, causing detention and alteration of cellular maturation [5, 10]; (2) many patients with Good’s syndrome are affected by opportunistic infections associated with cellular immunity defects, arising from the loss of memory or ‘naïve’ T CD4 cells [4, 5]; and (3) autoimmunity. Studies on paraneoplastic syndromes such as pure red cell aplasia with thymoma show that auto-antibodies or auto-reactive T lymphocytes could directly or indirectly inhibit erythropoiesis, so that the loss of B cell function could be secondary to the immune destruction [5].
The most common infection in these patients is recurrent pneumonia, secondary to encapsulated organisms (humoral response). They also develop opportunistic infections such as retinitis or colitis caused by cytomegalovirus, mucocutaneous infection caused by candida, pneumonia from Pneumocystis jiroveci, among others (cellular response) [4, 9].
Diarrhoea was reported in almost half of patients with Good’s syndrome. The mechanisms through which hypogammaglobulinaemia and thymoma cause diarrhoea are not very clear. It has been suggested that it could be related to vellositary atrophy, which could be resolved by re-establishment of the immunological state. On the other hand, patients are especially susceptible to gastrointestinal pathogens. Still, often no germ is identified. Among the cases in which it was possible to isolate a bacterial agent, enterobacterias were the most common pathogens. Other pathogens isolated were Giardia lamblia and cytomegalovirus [2, 4]
Treatment with immunoglobulin replacement has been reported in numerous cases of patients with Good’s syndrome, and around 38% show a decrease in the number of infections after treatment [2, 8]. This treatment is also associated with a decreased use of antibiotics and in the number of hospital admissions. Other forms of therapy reported in the literature include immunosuppressant treatment, splenectomy, and plasmapheresis [8, 13].
Autoimmune diseases such as pure red cell aplasia, myasthenia gravis, diabetes mellitus, ulcerative colitis, and haemolytic anaemia may occur in association with Good’s syndrome [2]. According to a systematic review by Kelesidis et al, pure red cell aplasia and myasthenia gravis are the autoimmune pathologies most commonly associated with Good’s syndrome, occurring in 34.8% and 15.7% of cases, respectively [8]. This association, as mentioned previously, suggests the potential role of autoantibodies or immunity by cells, in the pathogenesis of hypogammaglobulinaemia [5].
Pure red cell aplasia is often diagnosed soon after the diagnosis of thymoma. In the Mayo Clinic, the majority of patients are diagnosed within a month of being diagnosed with thymoma and the rest between four and 117 months after thymoma resection [11].
According to a review of the existing literature, treatment for pure red cell aplasia associated with thymoma is based on managing the base pathology, supportive transfusion therapy, and immunosuppressant treatment [11, 12, 14, 15].
In our case, the patient was diagnosed with Good’s syndrome and pure red cell aplasia two years after being diagnosed with thymoma in relation to a recurring tumour and in the context of inter-current infections associated with anaemic syndrome.
Currently, there are no solid data in the literature in relation to the treatment of patients with Good’s syndrome, pure red cell aplasia, and thymoma. With our patient, we decided to treat the recurrent base pathology with chemotherapy, intravenous gammaglobulin therapy, and transfusion support therapy as required.
Conclusion
The association of Good’s syndrome with pure red cell aplasia in the context of thymoma has been only scarcely reported in the literature. The presence of inter-current infections associated with severe anaemia with no clear aetiology should lead to a suspicion of this condition.
References
1. Tomaszek S, Wigle D and Keshavjee S (2009) Thymomas: review of current clinical practice Ann Thorac Surg 87 1973–80. DOI: 10.1016/j.athoracsur.2008.12.095 PMID: 19463649 PMCID: 2930778
2. Joven M, Palalay MP and Sonido C (2013) Case report and literature review on Good’s syndrome, a form of acquired immunodeficiency associated with thymomas Hawaii J Med Public Health 72 56–62 PMCID: PMC3585500
3. Ling-Ping C, Jui-Shan T and Wei-Ming L (2012) Myelodysplasia followed by Good’s syndrome: a unique manifestation associated with thymoma Kaohsiung J Med Sci 28 236–40 DOI: 10.1016/j.kjms.2011.10.012
4. Kelleher P and Misbah SA (2003) What is Good’s syndrome? Immunological abnormalities in patients with thymoma J Clin Pathol 56 12–6 DOI: 10.1136/jcp.56.1.12
5. Agarwal S and Cunningham-Rundles Ch (2007) Thymoma and immunodeficiency (Good syndrome): a report of 2 unusual cases and review of the literature Ann Allergy Asthma Immunol 98 185–90 DOI: 10.1016/S1081-1206(10)60695-3 PMID: 17304889 PMCID: 3102047
6. Bonilla F, Bernstein I and Khan D (2005) Practice parameter for the diagnosis and management of primary immunodeficiency Ann Allergy Asthma Immunol 94(suppl 1) S1–63 DOI: 10.1016/S1081-1206(10)61142-8 PMID: 15945566
7. Kitamura A, Takiguchi Y and Tochigi N (2009) Durable hypogammaglobulinemia associated with thymoma (Good syndrome) Inter Med 48 1749–52 DOI: 10.2169/internalmedicine.48.2375
8. Kelesidis T and Yang O (2010) Review: Good’s syndrome remains a mystery after 55 years: a systematic review of the scientific evidence Clin Immunol 135 347–63 DOI: 10.1016/j.clim.2010.01.006 PMID: 20149753
9. Tarr P, Sneller M and Mechanic L (2001) Infections in patients with immunodeficiency with thymoma (Good syndrome). Report of 5 cases and review of the literature Medicine 80 123–33 DOI: 10.1097/00005792-200103000-00005 PMID: 11307588
10. Oritani K, Medina KL and Tomiyama Y (2000) Limitin: an interferon-like cytokine that preferentially influences B-lymphocyte precursors Nat Med Immunol 3 659–66 DOI: 10.1038/76233
11. Thompson C and Steensma D (2006) Pure red cell aplasia associated with thymoma: clinical insights from a 50-year single-institution experience Br J Haematol 135 405–7 DOI: 10.1111/j.1365-2141.2006.06295.x PMID: 17032177
12. Chen-Sung L, Yuan-Bin Y and Han-Shui H (2009) Pure red cell aplasia and hypogammaglobulinemia in a patient with thymoma J Chin Med Assoc 72 34–8 DOI: 10.1016/S1726-4901(09)70017-6
13. Degos L, Faille A and Housset M (1982) Syndrome of neutrophil agranulocytosis, hypogammaglobulinemia, and thymoma Blood 60 968–72 PMID: 7115963
14. Masaoka A, Hashimoto T and Shibata K (1989) Thymomas associated with pure red cell aplasia: histologic and follow-up studies Cancer 64 1872–78 DOI: 10.1002/1097-0142(19891101)64:9<1872::AID-CNCR2820640920>3.0.CO;2-0 PMID: 2507126
15. Kuo T and Shih LY (2001) Histologic types of thymoma associated with pure red cell aplasia: a study of five cases including a composite tumour of organoidthymoma associated with an unusual lipofibroadenoma Int J Surg Pathol 9 29–35 DOI: 10.1177/106689690100900106 PMID: 11469342
16. Hermaszewski R and Webster A (1993) Primary hypogammaglobulinaemia: a survey of clinical manifestations and complications Q J Med 86 31–42 PMID: 8438047