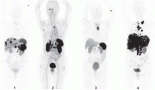Li–Fraumeni-associated pancreatic neuroendocrine tumour and XAF1 p.Glu134Ter risk modifier variant
Rachel P Riechelmann1, Diogo C Soares2, Carla Dias1,3, Dirce M Carraro4,5 and Giovana T Torrezan4,5
1Department of Clinical Oncology, A.C. Camargo Cancer Center, Rua Antônio Prudente 211, São Paulo, SP 01509‐010, Brazil
2Department of Oncogenetics, A.C. Camargo Cancer Center, Rua Antônio Prudente 211, São Paulo, SP 01509‐010, Brazil
3Instituto do Câncer do Estado de São Paulo, São Paulo, SP 01246-000, Brazil
4Genomics and Molecular Biology Group, International Research Center/CIPE, A.C. Camargo Cancer Center, Rua Antônio Prudente 211, São Paulo, SP 01509‐010, Brazil
5National Institute of Science and Technology in Oncogenomics and Therapeutic Innovation, São Paulo, SP 01509‐010, Brazil
Abstract
Studies have demonstrated that up to 17% of patients with pancreatic neuroendocrine tumours (pNETs) present pathogenic germline variants (PGVs) in several different genes, irrespective of family cancer history. Li–Fraumeni syndrome (LFS) is an autosomal dominant cancer predisposition syndrome related to PGVs in the TP53 gene. A previous case of a pNET associated with LFS (c.1009C > T, p.R337C) has been reported. Here we report the first case of a patient with pNET and TP53 p.R337H and XAF1 p.E134* germline variants, expanding the knowledge of LFS and germline mutations in neuroendocrine tumours.
Keywords: Li–Fraumeni, neuroendocrine tumour, pancreas neoplasm
Correspondence to: Rachel P Riechelmann
Email: rachel.riechelmann@accamargo.org.br
Published: 08/12/2022
Received: 18/07/2022
Publication costs for this article were supported by ecancer (UK Charity number 1176307).
Copyright: © the authors; licensee ecancermedicalscience. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Introduction
Hereditary genetic syndromes associated with pancreatic neuroendocrine tumours (pNETs) are rare, occurring in nearly 10% of cases, mostly in multiple endocrine neoplasia type 1 (MEN1), von Hippel–Lindau disease (VHL), neurofibromatosis type 1 or tuberous sclerosis complex 1 and 2 (TSC1 or TSC2). Yet recent studies have shown that monoallelic pathogenic germline variants (PGVs) have been found in 16%–17% of pNET patients, including PGVs in MUTYH, CHEK2 and BRCA2, without known classic clinical criteria for suspecting hereditary syndromes [1–3].
Li–Fraumeni syndrome (LFS) is an autosomal dominant cancer predisposition syndrome related to PGVs in TP53 gene. Several studies have demonstrated a great variety of tumours that arise in individuals with LFS, such as breast cancer, bone and soft tissue sarcomas, adrenocortical carcinomas and central nervous system tumours. Less commonly, LFS patients may develop other tumours such as colorectal and gastric adenocarcinomas, haematological malignancies, thyroid cancer, melanoma, lung and prostate cancers. The majority of TP53 PGVs are located in p53 DNA-binding domain with patients presenting a highly penetrant phenotype. There is a founder variant c.1010G>A, p.(Arg337His) located in the p53 oligomerisation domain, highly prevalent in Southern and Southeastern Brazil and which is characterised by a wide range of malignancies but with a lower penetrance when compared to other PGVs (15%–20% versus 50% in p.R337H versus classical mutations carriers, respectively, develop cancer by 30 years of age); p.R337H carriers present a higher risk of developing adrenocortical carcinomas, renal tumours, papillary thyroid cancer and lung adenocarcinomas [4]. Interestingly, family cancer histories of carriers of the Brazilian founder TP53 p.R337H allele may present from isolated cancer cases to those with multiple tumours resembling classic LFS [4]. Recent studies have suggested that a germline nonsense variant E134*/Glu134Ter/rs146752602 in the tumour-suppressor gene X-linked inhibitor of apoptosis (XIAP)-associated factor 1 (XAF1) is a genetic modifier of p53 function and may induce a more aggressive cancer phenotype in LFS carriers of TP53 p.R337H [5].
Only one previous case of a pNET associated with LFS has been reported. The case was of a 43-year-old female with a G2 pNET and a rare TP53 PGVs on the residue 337, c.1009C > T, p.R337C, which was also found in her pNET tissue with an allele frequency of 93% [6]. Here we report the first case of a patient with pNET and TP53 p.R337H and XAF1 p.E134* germline variants.
Case report
The patient is a 60-years old previously healthy and asymptomatic male who underwent several laboratory and imaging tests to screen out cancer after his father was diagnosed with LFS (PGV in TP53, c.1010G>A, p.(Arg337His)). His family cancer history was significant: father with lung adenocarcinoma at the age of 52 years old (died with 52 years), brother with a central nervous system tumour at 19 years (died when 37 years old), brother with prostate cancer at 57 years (alive with 63 years), paternal uncle with oesophageal cancer at 78 years (died with 78 years), paternal cousin with breast cancer at 59 years (alive with 63 years), paternal uncle with soft tissue sarcoma at 62 years (died at 66 years), mother with gastrointestinal cancer at 47 years (died at 87 years), maternal uncle with central nervous system tumour at 42 years (died with 42 years), both grandmothers died with more than 70 years and without cancer history. Figure 1 describes his genogram.
An abdominal ultrasound was performed and identified multiple hypoechoid nodules of varying size, with the largest ones being of 28 × 24 mm and 26 × 18 mm located in segment IV. A magnetic resonance image of the abdomen showed a 3.5 × 2.7 cm nodular lesion in the pancreatic tail and numerous bilobar hepatic lesions measuring up to 5 cm. A liver biopsy was performed in November 2020, of which the pathology showed a well-differentiated grade 3 neuroendocrine tumour infiltrating the liver tissue; tumour cells stained positive by immunohistochemistry for chromogranin A, synaptophysin, CD56 and alpha-antitrypsin, with a ki67 proliferative index of 35%. The 68Gallium-DOTATATE positron emission tomography/computed tomography (PETCT) showed positive uptake in all tumour lesions: a pancreatic tail lesion with a standardised uptake value (SUV) of 67.3 and multiple hepatic nodules with SUV maximum of 47. Other imaging and laboratory tests performed to screen out cancer in individuals with LFS did not identified other neoplasms.
Genetic evaluation was undertaken to confirm the TP53 p.R337H germline variant and to investigate loss of heterozygosis (LOH) of TP53 wild allele in the tumour tissue. We found heterozygous PGVs in TP53 (c.1010G>A, p.[Arg337His]) and in XAF1 (c.400G>T, p.[Glu134Ter]). Tumour DNA extracted from a paraffin-embedded biopsy of a pNET liver metastasis showed loss of the wild-type alternate allele (LOH) of both germline variants (Figure 2), with allele frequency of 92% for TP53 and allele frequency of 91% for XAF1.
Because the patient was asymptomatic and presented pNET lesions with high SUV in the 68Gallium-DOTATE PETCT, we started treatment with octreotide 30 mg long-acting release (LAR) monthly in January 2021. Re-imaging in 4 months demonstrated high-volume progression in liver metastases, weight loss and abdominal pain. Modified FOLFIRINOX was started, and the patient experienced significant clinical benefit after 2 months of treatment: he gained weight, had complete resolution of his abdominal discomfort and radiological imaging reported overall disease stabilisation with minor tumour shrinkage.
Informed consent was provided by the patient to report the case and perform genetic analyses.
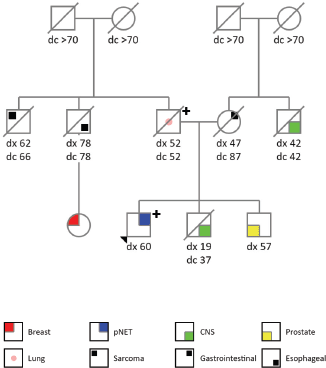
Figure 1. Genogram.
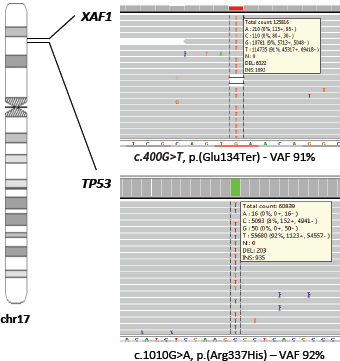
Figure 2. Loss of the wild-type alternate allele (LOH) of TP53 and XAF1 germline variants.
Discussion
We report the first pNET case associated with p.Glu134Ter XAF1 and p.R337H TP53 variants, documented by LOH in the tumour. This represents a new discovery that expands the clinical spectrum of LFS, particularly for the founder variant LFS highly prevalent in Southern and Southeastern of Brazil.
PGVs in TP53 gene detected in patients with classical LFS most commonly occur within the DNA-binding domain, leading to the production of a dysfunctional p53 protein [7]. While the oligomerisation domain TP53 p.R337H variant encountered in our patient is relatively uncommon worldwide, it is predominantly found in Southern and Southeastern of Brazil, where it occurs in a frequency of 0.03% [8, 9]. Brazilian families with LFS often carry this founder TP53 germline variant that features a guanine to adenine transition and a subsequent arginine to histidine replacement (c.1010G > A, p.R337H), which confers higher risk of several tumours, specially adrenocortical carcinoma [4]. A recent study proposed that a XAF1 germline variant, p.E134*, may influence cancer risk and phenotype among p.R337H carriers, given it functions as a tumour suppressor gene that operates in a positive feedback loop with p53 [10]. Pinto et al [5] identified that 69% of p.R337H carriers also have the XAF1 p.E134* variant in the same haplotype, being enriched in patients with cancer, conferring higher risks of sarcoma and multiple primary tumours. Whether the cosegregation of TP53 and XAF1 led to the late onset of a G3 pNET in our patient is unknown.
To the authors’ knowledge, there is only one pNET case report associated with LFS, albeit with a different genotype. A 43-year-old Russian and non-Ashkenazi Jewish descendent female with a localised G2 pNET presented a large pancreatic mass (largest diameter was 12.7 cm) which was surgically removed and pathologically diagnosed as a well-differentiated pNET with a ki67 index of 3.5% and a mitotic rate of less than 1 in 50 high-powered fields [6]. Concurrently, the patient was diagnosed with a 6 cm heterogeneous lesion consistent with an angiomyolipoma in her left kidney. During follow-up, she developed a choroid plexus papilloma which was resected. She had a 7-year-old daughter who died from a grade 3 brain astrocytoma with a TP53 somatic mutation identified in the tumour tissue. The patient was found to carry a pathogenic germline TP53 variant, c.1009C > T, p.R337C, resulting in arginine to cysteine substitution, which was also present in her pNET tissue with an allele frequency of 93%, strongly suggesting LOH.
Considering Knudson’s two-hit hypothesis, verifying the presence of biallelic inactivation (e.g. inferring LOH by tumour sequencing) can help establish whether PGVs in tumour suppressor genes contributed to cancer development. Both our case and the previously described LFS-associated pNET showed loss of the wild-type TP53 allele, confirming the biallelic TP53 inactivation in the tumours. Although only two cases are not enough to confirm a causal association, it is a compelling finding.
Our case report has clinical utility. While the Chompret criteria (e.g. the clinical criteria used to identify suspected LFS individuals who are candidates for genetic testing) does not include the investigation of NET, for individuals with known LFS, routine surveillance with whole body magnetic resonance is probably sufficient to screen for several cancer types, including pNET [6]. Additionally, two prospective cohorts conducted by our group have documented that pNET patients may present PGVs in up to 17% of cases, including BRCA2, CHEK2, XPC, MUTYH, MEN1, VHL and TSC2 [1–3] and now, TP53 p.R337. We think patients with early onset pNET, regardless of cancer family history, should be considered for the investigation of PGVs. Such investigations could have tremendous impact on the diagnosis and surveillance of cancer predisposing syndromes, for the pNET patient him/herself, as well as for his/her family members.
Several questions remain: the true penetrance of pNET in TP53 p.R337H and XAF1 p.E134* carriers, the role of PGVs in TP53 residue 337 and pNET onset (both pNET cases developed in carriers of alterations in this amino acid), whether other NETs may arise in this context, the role of XAF1 mutations as a genetic modifier of TP53-R337H/C carriers in the development of pNET, the biological behaviour of LFS-associated pNET and its sensibility to standard NET-directed drugs.
Conclusion
In conclusion, we report the first case of a pNET in a TP53 p.R337H and XAF1 p.E134* carrier. Our case expands on the knowledge of the heterogeneity of LFS, specifically among carriers of the TP53 p.R337H mutation.
Conflicts of interest
None to declare.
Funding
This research was funded by The São Paulo Research Foundation (FAPESP grant numbers 2014/50943-1 and 2018/06269-5), National Council for Scientific and Technological Development (CNPq grant number 465682/2014-6) and Coordination for the Improvement of Higher Education Personnel (CAPES grant number 88887.136405/2017-00).
References
1. Scarpa A, Chang DK, and Nones K, et al (2017) Whole-genome landscape of pancreatic neuroendocrine tumours Nature 543(7643) 65–71 https://doi.org/10.1038/nature21063 PMID: 28199314
2. Raj N, Shah R, and Stadler Z, et al (2018) Real-time genomic characterization of metastatic pancreatic neuroendocrine tumors has prognostic implications and identifies potential germline actionability JCO Precis Oncol 2 1–8
3. Riechelmann R, de Paula C, and Donadio M, et al (2020) Young adults with neuroendocrine tumors present a high rate of pathogenic or likely pathogenic germline variants in cancer predisposing genes Ann Oncol 31 236–237 Date accessed: 01/06/20 https://doi.org/10.1016/j.annonc.2020.04.067
4. Achatz MI and Zambetti GP (2016) The inherited p53 mutation in the Brazilian population Cold Spring Harb Perspect Med 6(12) a026195 https://doi.org/10.1101/cshperspect.a026195 PMID: 27663983 PMCID: 5131754
5. Pinto EM, Figueiredo BC, and Chen W, et al (2020) XAF1 as a modifier of p53 function and cancer susceptibility Sci Adv 6(26) eaba3231 https://doi.org/10.1126/sciadv.aba3231 PMID: 32637605 PMCID: 7314530
6. Aversa JG, De Abreu FB, and Yano S, et al (2020) The first pancreatic neuroendocrine tumor in Li-Fraumeni syndrome: a case report BMC Cancer 20(1) 256 https://doi.org/10.1186/s12885-020-06723-6 PMID: 32228502 PMCID: 7106707
7. Luca JW, Strong LC, and Hansen MF (1998) A germline missense mutation R337C in exon 10 of the human p53 gene Hum Mutat 11 S58–61 https://doi.org/10.1002/humu.1380110121
8. Custódio G, Parise GA, and Kiesel Filho N, et al (2013) Impact of neonatal screening and surveillance for the TP53 R337H mutation on early detection of childhood adrenocortical tumors J Clin Oncol 31(20) 2619–2626 https://doi.org/10.1200/JCO.2012.46.3711 PMID: 23733769 PMCID: 3808236
9. Seidinger AL, Caminha IP, and Mastellaro MJ, et al (2020) TP53 p.Arg337His geographic distribution correlates with adrenocortical tumor occurrence Mol Genet Genomic Med 8(9) e1168 https://doi.org/10.1002/mgg3.1168 PMID: 32592449 PMCID: 7503091
10. Lee MG, Han J, Jeong SI, et al (2014) XAF1 directs apoptotic switch of p53 signaling through activation of HIPK2 and ZNF313 111(43) 15532–15537 PMID: 25313037 PMCID: 4217407