Therapeutic strategies of recurrent glioblastoma and its molecular pathways ‘Lock up the beast’
Shaimaa M El-khayat1 and Waleed O Arafat2
1Cancer Management and Research Department, Medical Research Institute, Alexandria University, Alexandria 21568, Egypt
2Alexandria Clinical Oncology Department, Alexandria University, Alexandria 21568, Egypt
Abstract
Glioblastoma multiforme (GBM) has a poor prognosis—despite aggressive primary treatment composed of surgery, radiotherapy and chemotherapy, median survival is still around 15 months. It starts to grow again after a year of treatment and eventually nothing is effective at this stage. Recurrent GBM is one of the most disappointing fields for researchers in which their efforts have gained no benefit for patients. They were directed for a long time towards understanding the molecular basis that leads to the development of GBM. It is now known that GBM is a heterogeneous disease and resistance comes mainly from the regrowth of malignant cells after eradicating specific clones by targeted treatment. Epidermal growth factor receptor, platelet derived growth factor receptor, vascular endothelial growth factor receptor are known to be highly active in primary and recurrent GBM through different underlying pathways, despite this bevacizumab is the only Food and Drug Administration (FDA) approved drug for recurrent GBM. Immunotherapy is another important promising modality of treatment of GBM, after proper understanding of the microenvironment of the tumour and overcoming the reasons that historically stigmatise GBM as an ‘immunologically cold tumour’. Radiotherapy can augment the effect of immunotherapy by different mechanisms. Also, dual immunotherapy which targets immune pathways at different stages and through different receptors further enhances immune stimulation against GBM. Delivery of pro-drugs to be activated at the tumour site and suicidal genes by gene therapy using different vectors shows promising results. Despite using neurotropic viral vectors specifically targeting glial cells (which are the cells of origin of GBM), no significant improvement of overall-survival has been seen as yet. Non-viral vectors ‘polymeric and non-polymeric’ show significant tumour shrinkage in pre-clinical trials and now at early-stage clinical trials. To this end, in this review, we aim to study the possible role of different molecular pathways that are involved in GBM’s recurrence, we will also review the most relevant and recent clinical experience with targeted treatments and immunotherapies. We will discuss trials utilised tyrosine receptor kinase inhibitors, immunotherapy and gene therapy in recurrent GBM pointing to the causes of potential disappointing preliminary results of some of them. Additionally, we are suggesting a possible future treatment based on recent successful clinical data that could alter the outcome for GBM patients.
Keywords: glioblastoma, GBM, recurrent GBM, recurrent high-grade glioma
Correspondence to: Waleed O Arafa
Email: warafat@alexmed.edu.eg
Published: 22/01/2021
Received: 05/10/2020
Publication costs for this article were supported by ecancer (UK Charity number 1176307).
Copyright: © the authors; licensee ecancermedicalscience. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Introduction
A group of malignant brain tumours called ‘Gliomas’ arises from the supporting neuroglial cells (astrocytic or oligodendroglial cells). Although current WHO classification includes epigenetic, genetic and clinicopathological features of brain tumours, most clinicians still divide brain tumours into low-grade glioma including (Grades I, II) and high-grade glioma (HGG) (grades III, IV) [1]. Glioblastoma multiforme (GBM) WHO grade IV is the most aggressive and commonest of these brain tumours and constitutes up to 54% of all gliomas and 16% of all intracranial tumours (primary and metastatic) [2]. Despite researchers’ efforts to improve the outcome of patients with GBM, the only approved treatments are still maximum safe resection followed by radio-chemotherapy with alkylating agent temozolomide (TMZ) then adjuvant TMZ with tumour-treating fields [3], this initial stage of treatment takes around 9 months [4–6], and its result in 7–9 months progression-free survival (PFS) and 15 months overall survival (OS) [7]. However, 100% of GBM will recur and response to subsequent treatment is very minimal [8], that is why we need to understand the molecular basis that led to recurrence and possible targeted treatments that can be developed to overcome its resistance to almost all treatments in the recurrent setting.
As suggested by the term ‘multiforme’, GBM is characterised by intra-tumour heterogeneity not only on cellular but also on molecular levels [9]. This heterogeneity is one of the principal reasons for therapeutic resistance and recurrence [9]. It is believed that this happens due to the biological selection of resistant malignant clones and then they acquire genetic alterations making them more aggressive after primary treatment [10]. One of the famous publications in which scientists extensively studied GBM at the molecular level was the cancer genome atlas (TCGA). TCGA has offered insights into the genetic regulation of GBM by a molecular GBM classification with the identification of molecular subgroups with prognostic significance [11, 12]. The four subgroups of GBM are classical, neural, pro-neural and mesenchymal as shown in Figure 1, pointing to the defective molecular pathway in each sub-group which could help to develop specific targeted treatments in the future and better designing of clinical trials on a molecular basis [12].
Till now, no clinical reflection of the four subgroups with only a slight survival advantage to the pro-neural subtype [13, 14].
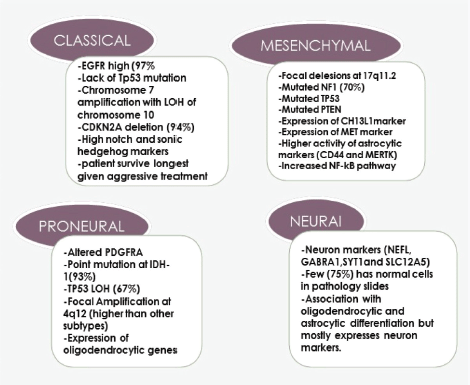
Figure 1. Four subtypes of GBM and the dominant genes and molecular abnormality in each group.
This review article considers the major therapeutic strategies currently being investigated in the field of recurrent GBM, focusing on approaches with not only pre-clinical but also clinical data. We aim to discuss novel and experimental tyrosine kinase receptor inhibitors, immunotherapy and gene therapy pointing to the underlying pathways that lead to their promising role in recurrent GBM. We also added a final section on the most important future direction that scientists are trying to apply to treat recurrent GBM based on pre-clinical data to improve the outcome of these patients.
Methods
We searched the MEDLINE, PubMed database for high impact factor journals at least 1.8 with high citations. We also searched clinical trials.gov for phase I, II trials with reported PFS, OS published on tyrosine receptor kinase inhibitors, immunotherapy and gene therapy in recurrent GBM and discussed them. Ongoing trials and pre-clinical relevant up-to-date studies related to this subject have also been discussed. We used in our search the keywords glioblastoma, GBM, recurrent GBM and recurrent high-grade glioma.
Discussion
Receptor tyrosine kinase (RTK)
The RTK inhibitors are one of the most extensively studied drugs in oncology, we will discuss in the following section four of the main growth factor receptors in GBM and targeted treatments against them focusing on the applied clinical experience from clinical trials.
Epidermal growth factor receptor (EGFR)
Human EGFR (EGFR; HER-1) is over-expressed in 40%–60% of primary GBM tumours and mostly occurs in the classical subtype (Figure 1) [15], but EGFR mutation which leads to EGFRvIII expression (Figure 2) present in 20%–30% of primary GBM [15]. In this regard, a study of 186 pairs of primary and recurrent GBM samples found that patients with recurrent glioblastoma multiform (rGBM) do not represent specific molecular subtypes and almost 80% of recurrent diseases retain the same molecular abnormalities as in the primary tumour samples [16]. Therefore, scientists designed clinical trials investigating targeted treatment for recurrent GBM based on an almost similar percent of activating mutations in primary and recurrent samples. Although the EGFR gene is commonly amplified in GBM, this does not correlate with responsiveness to EGFR inhibitor in most of the trials. The mutations of EGFR in GBM is linked to ‘gain-of-function miss-sense mutations or in-frame deletions affecting the extracellular domain’ [17, 18]. EGFRvIII is always active regardless of the presence of ligand or not with dysregulated downstream pathways [19]. This mutant ligand-independent pathway is believed to create a state of ‘pathway addiction’ in which the tumour dies if debriefed from this signal by tyrosine kinase inhibitors (TKIs) [20].
EGFR receptor is activated by two mechanisms as mentioned above, either by receptor over-expression or multiple ligand-independent and ligand-dependent pathways which will lead to stimulation of subsequent downstream mitogen-activated protein kinase, phosphatidylinositol-3-OH kinase (PI3K) and Src kinase pathway besides signal transducers and activators of transcription (STAT) transcription factor activation [21]. These events starting from upstream mutation will lead to an intracellular cascade of events leading to gene transcription, cell proliferation, survival, invasion and angiogenesis (Figure 2).
Sometimes phosphatase and TENsin homolog (PTEN) activity is lost which mainly acts as a tumour suppressor protein that inhibits the PI3K pathway, in this situation, there will be resistance to EGFR kinase inhibitors [22]. Patients with PTEN-deficient tumours could benefit from downstream inhibition of the PI3K pathway, maybe at the level of the mammalian target of rapamycin (mTOR), with EGFR inhibitors.
EGFR TKIs are classified into three main categories: first-generation inhibitors that target EGFR and its co-receptor HER2 and bind to it reversibly like (gefitinib, erlotinib and lapatinib); second-generation irreversible inhibitors (afatinib, neratinib and dacomitinib) and third-generation TKIs.
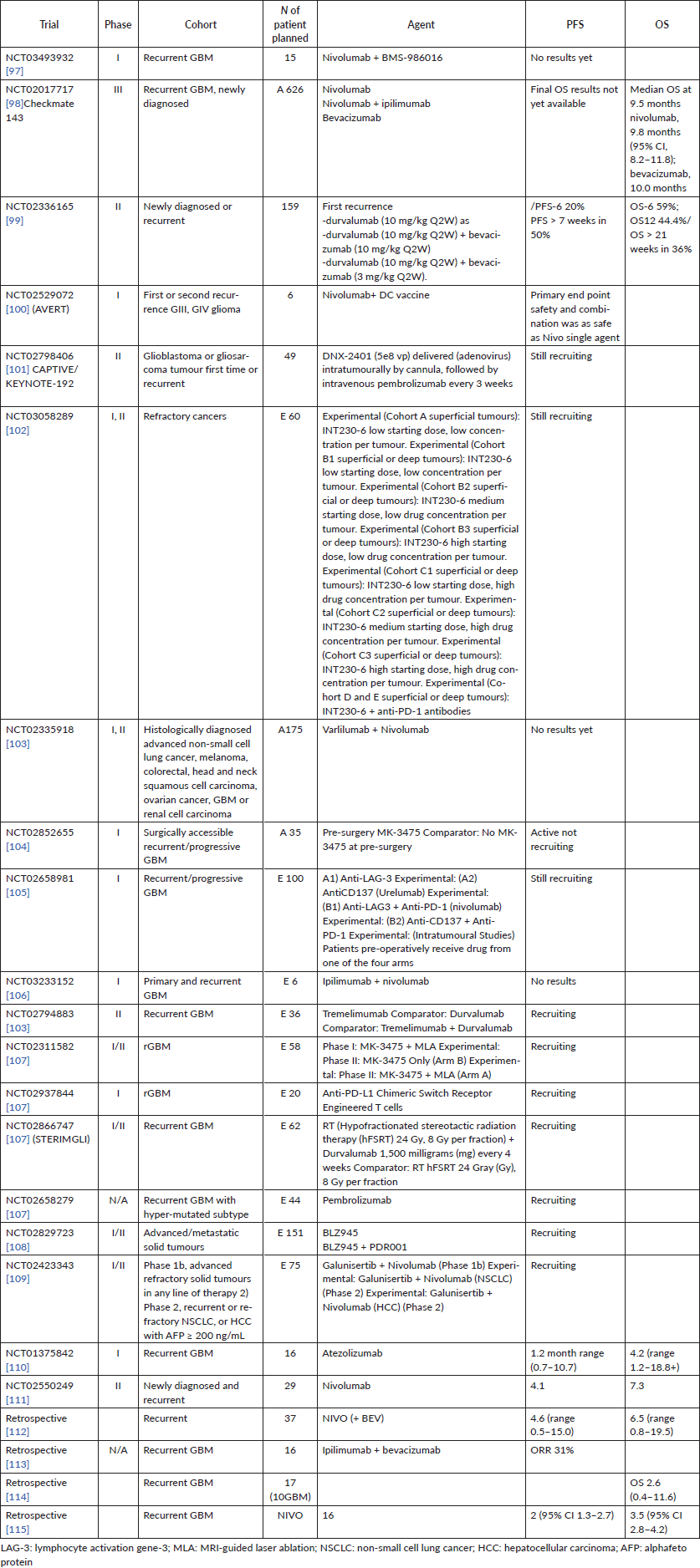
Almost all trials for recurrent GBM patients ‘based on a high percentage of them expressing EGFR (40%–60%)’ evaluating EGFR TKIs utilised a continuous daily dosing schedule but also included unenriched participants (Table 1). Some studies evaluated TKI monotherapy, others evaluated the combination regimen.
Gefitinib was evaluated in phase II single-arm trial that included 57 patients during the first recurrence and found that no radiological response was found in them, PFS-6 (PFS at 6 months) was 13% and OS was 39.4 weeks. There was no correlation between EGFRvIII mutation’s presence or absence of EGFR over-expression with the outcome [23]. Another study that evaluated gefitinib in the neo-adjuvant setting found that its concentration at the tumour was 20 times more than that found in the plasma but this finding was not associated with downstream pathway inhibition. So the drug acts effectively on the EGFR receptor upwards, but no effect on downstream pathways, this is also observed with erlotinib [24] and lapatinib. These studies suggest that probably first generation EGFR TKIs do not inhibit the EGFR signalling in GBM effectively and the above-mentioned observation could be the reason for the failure of these drugs till now.
Erlotinib was tested on 44 recurrent GBM patients and again the radiological response was not observed and PFS-6 was 3% [25]. Later studies proved that erlotinib has poor central nervous system (CNS) penetration due to interaction with P-gp efflux transporter and breast cancer resistance protein [26]. Other study which compared erlotinib with chemotherapy at first recurrence found that the outcome was comparable in both arms and EGFRvIII mutation was linked to poor PFS [27].
Resulting from the hypothesis of thinking of the possibility that stimulation of downstream pathways or activation of other survival pathways may cause EGFR resistance, subsequent studies evaluated the combination of EGFR TKI with agents that inhibit these signalling pathways [28]. Many studies evaluated EGFR TKI combined with (mTOR) inhibitors that act as a mediator of the PI3K/AKT phosphatidylinositol-3 kinases/AKT, also known as protein kinase B (PKB), signalling pathway. Patients with recurrent GBM were evaluated in a phase I trial to determine the maximum tolerable dose (MTD) of gefitinib with (an oral mTOR inhibitor) and reported PFS-6 of 23.5% [29]. Then another phase II study which was a single-arm on 32 heavily pretreated, recurrent GBM patients evaluated combining erlotinib with sirolimus and found that no radiological response was found and PFS-6 was only 3.1% [30], and also, unfortunately, no correlation between OS and EGFRvIII, pEGFR and EGFR amplification.
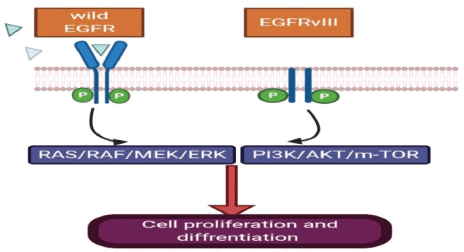
Figure 2. EGFRvIII has an extracellular domain truncation from exons 2 to 7, which results in the deletion of amino acids 6-273 and renders the mutant receptor incapable of binding the ligand. EGFRvIII can display constitutively active signalling independent of ligand.
Some of the finished and ongoing trials of anti-EGFR in rGBM are demonstrated in Table 1;
Depending on the pre-clinical findings that VEGF signal activation acts as a mediator of EGFR resistance, a combination of EGFR inhibitor and VEGF2R2 inhibitors was introduced to clinical trials to be tested [38, 39]. Phase II study evaluated erlotinib plus bevacizumab ‘a humanised monoclonal antibody against VEGF that is Food and Drug Administration (FDA)-approved for recurrent GBM’ [40] was conducted on patients with rGBM [41]. Erlotinib was administered at 200 mg/day and 500 mg/day for participants taking and not taking enzyme-inducing antiepileptic drugs, respectively, and bevacizumab was administered at 10 mg/kg biweekly. In the above study, PFS-6 was 28% and OS was 42 weeks, and these results were the same as that found with bevacizumab monotherapy.
Table 1. Some phase I, II trials of anti-EGFR in recurrent GBM and the status of each trial, whether finished or still ongoing.
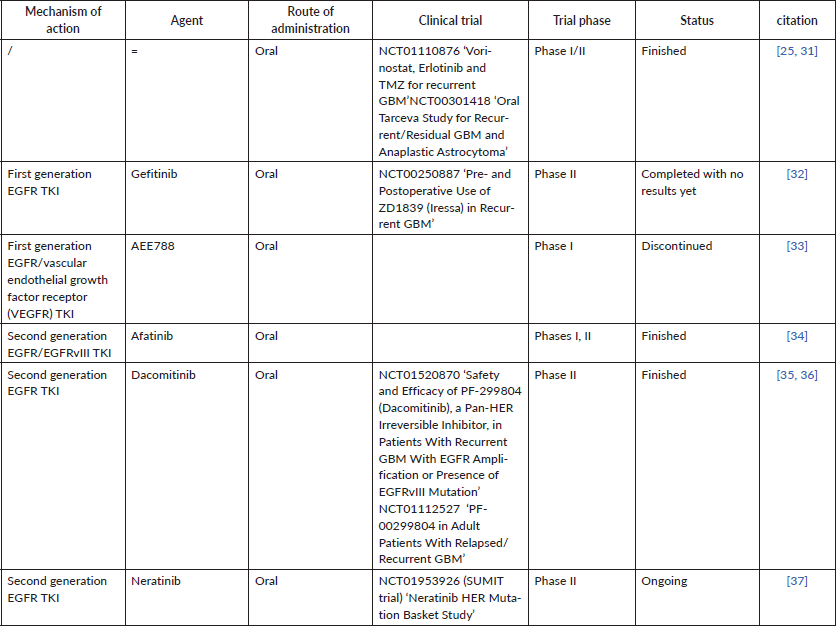
As regards trials that evaluated second-generation EGFR, in phase I/II study of afatinib with or without protracted TMZ in patients with GBM after recurrence, provided that all patients received standard chemo-radiotherapy with TMZ at presentation [42]. The phase I results of the trial found that MTD of afatinib with TMZ is 40 mg/day for afatinib and 75 mg/m2/day for TMZ when combined. And the phase II results of the same trial found that the PFS-6 for the afatinib monotherapy arm, afatinib-protracted TMZ arm and combination arm was 3%, 23% and 10%, respectively. Different biomarkers were evaluated like EGFR, EGFRvIII, PTEN, pAKT and O6-methylguanine-DNAmethyltransferase (MGMT) but none of them correlated with the outcome although a non-significant association with EGFRvIII expression and better outcome were observed in patients treated with afatinib.
Dacomitinib single agent was evaluated in two phases II studies [43, 44]. The first one included two cohorts; one of them included patients with EGFR over-expression with no EGFRvIII mutation and achieved PFS6 of 13.3% and OS of 7.8 months, the second cohort included patients with EGFR over-expression and EGFRvIII mutation and PFS6 was 5.9% and OS of 6.7 months. The two cohorts received dacomitinib till disease progression or unacceptable side effects [35]. The second study contains three arms and still ongoing, one of the three arms is giving dacomitinib as a neoadjuvant treatment before surgery and this will help to determine the penetration of dacomitinib to the blood–brain barrier and also its capability of inhibiting intra-tumour phosphorylation. The other two arms include patients who are naïve and previously exposed to bevacizumab. Neratinib is also under evaluation in tumours with either EGFR mutation or amplification in phase II study [45].
Platelet derived growth factor receptor (PDGFR)
PDGFR is another member of the TKI family and is overexpressed in HGGs, especially in GBs [46]. Platelet derived growth factor receptor A (PDGFRA) is over-expressed in about 15% of GBMs [47]. That is why researchers make efforts to target this receptor and its pathway. Pre-clinical studies are undergoing to test PDGFR inhibitors in vivo and in vitro and some of these inhibitors are approved for clinical trials. Imatinib (Gleevec) is one of these drugs which has an inhibitory effect on PDGFR. Although imatinib has activity in other malignancies, it did not show significant activity in HGG especially GBM in the recurrent settings. The tumour growth and OS remained unchanged whether it was used as a single agent or in combination with hydroxyurea [48, 49]. Recently, in vitro studies on GB cells found that imatinib increases the migration and invasion of GB cells, a fact that explains the previous failures of the drug [50].
Tandutinib is another platelet derived growth factor receptor B (PDGFRB) inhibitor, which was evaluated in clinical trials in recurrent GBM and was found to have a little effect [51]. AG1433 is also another PDGFR inhibitory molecule that proved activity in pre-clinical trials in several in vitro HGG cell lines [6]. In 2019, it was tested on 11 and 15 HGG cell lines with radiotherapy or not, and found that AG1433 was effective and adding radiation to it does not increase its activity [52] (Figure 3).
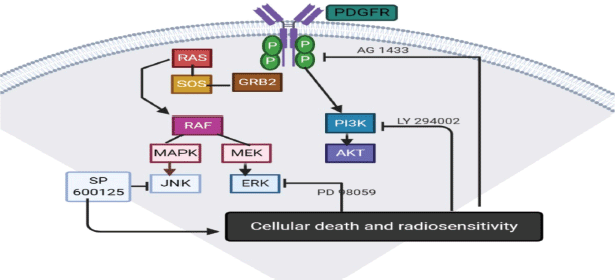
Figure 3. PDGFR pathway.
Vascular endothelial growth factor receptor (VEGFR)
GBM is one of the highly vascularised tumours and increased microvasculature is one of its hallmarks of pathology [53]. Many previous studies focused on targeting angiogenesis which occurs by spurting of new capillaries and blood vessels and by also recruitment of endothelial cells to provide blood supply for the growing tumour [54]. VEGFR especially VEGFR2 is an important target in glioblastoma. Vatalanib (PTK787) is a VEGFR2, PDGFR and c-kit inhibitor, scientists found that it has a little effect on GBM alone with either chemotherapy or radiotherapy [55]. In phase II study, Sorafenib which is another VEGFR inhibitor with temsirolimus had a small effect on GBM [56]. Despite that tivozanib ‘an inhibitor of angiogenesis’ had good anti-angiogenic effects on GB, it failed to change the tumours’ volume [57]. Pazopanib also was combined with lapatinib but the results were disappointing [58]. Cediranib, ‘a small molecule inhibitor of VEGFR, PDGFR and c-kit’, showed a small improvement in the neurological status of the patients but did not change PFS or OS [59]. SU1498 is another VEGFR inhibitor that has limited activity on GBM [60].
Recent data suggest that YKL-40 is a good marker for angiogenesis in recurrent GBM for which targeted treatment may improve the outcome. YKL-40 is a mesenchymal marker which is named as ‘human cartilage glycoprotein-39 or chitinase-like protein-1’ and probably has an important role in migration and motility of glioma stem cells (GSCs) and their differentiation into endothelial cells, that is why it has a role in angiogenesis [61]. It was proven that YKL-40 causes up-regulation of VEGF expression and new tumour vasculature induced by YKL-40 is partially dependent on VEGF [62]; therefore, targeted treatments against YKL-40 could affect GB’s treatment.
Fibroblast growth factor receptor (FGFR)
Although FGFR mutations are not frequent in GBM, several studies suggest that modification of the FGFR signalling pathway stimulates GBM progression and patient survival [63]. Small molecules that inhibit the FGFR TKI are under investigation [64]. Some of them are non-specific to FGFR and act on other RTK like ‘lenvatinib, ponatinib, dovitinib and brivanib’, and others selectively target FGFR, like PD173074, BGJ398, AZ4547 and JNJ-493 [65]. A study, which used a large-scale shRNA to know the FGFR signalling to be targeted in glioma at the paediatric age, found that dovitinib, ponatinib, PD173074 and AZ4547 can inhibit the growth of glioma cells in vitro more than TMZ [66]. In December 2019, a trial involving BGJ398 in rGBM was completed, but no results have been published yet. Another phase I/II trial using TAS-120 is currently recruiting patients with metastatic solid tumours, regardless of fibroblast growth factor (FGF)/FGFR-related abnormalities [67].
Multi-target tyrosine receptor kinase inhibitors (TRKI) agents
The multi-targeted approaches may represent a good choice for effective selection of resistant tumour subtypes. Vandetanib is one of these multitargeted TKI (VEGFR, EGFR) that was evaluated in clinical trials for patients with recurrent GBM. The drug was safe and tolerable but its antitumour effects were limited [68].
Another phase I clinical trial proved the safety of administrating vandetanib in combination with sirolimus in patients with rGBM [69]. Two other multitarget (cabozantinib) and (PD173074) are small molecules that act by inhibiting VEGFR and other receptors. Cabozantinib achieved good results in vitro and clinical trials, and PD173074 had good results in vitro. Sunitinib is a multi-kinase inhibitor of VEGFR, PDGFR, fms-related receptor tyrosine kinase 1 (FLT1), FLT1/kinase insert domain receptor (KDR), FLT3 and Ret Proto-Oncogene (RET) kinases with no encouraging results in patients with GBM [70].
Immunotherapy in GBM
Immunotherapy stimulates the immune system to recognise, target and get rid of tumour cells. Many trials have focused on developing immunomodulating therapies to restore the functional ability of different immune cells against neoplastic cells and with promising results in several tumour types including melanoma, lung cancer, urothelial tumours and colon cancer [71]. Recent efforts and advances in translational research have led promising several strategies for immunomodulation in GBM, we discussed here the potential limitations and advances of immunotherapy in GBM. First limitations of immunotherapy in GBM is based on that immunotherapy has been most successful in tumours with high tumour mutational burden (TMB) but have yet to yield breakthroughs in GBM. GBM has low TMB despite its profound heterogeneity, rendering it an intrinsically immunologically quiet disease. Further suggested limitations of immunotherapy in GBM include the immunosuppressive environment that down regulates antigen presentation and disengages infiltrating immune cells [72, 73]. Also, immunotherapies can lead to inflammation within the intracranial space which could result in severe treatment-limiting neurological complications due to increased vasogenic oedema, autoimmune encephalitis and cytokine release syndrome [74, 75].
To leverage the natural immune response and restore its elimination ability of glioblastoma malignant cells, crucial understanding of the BBB and the tumour microenvironment and its complex interaction with the immune system is required In GBM, the BBB integrity is changed due to endothelial tight junctions damage reflecting molecular composition changes [76]. The BBB breakdown allows CD8 T cells to migrate to the CNS, and stimulation of the innate and adaptive immune responses which produce cytokines and chemokines to recruit lymphocytes and up-regulate immune-modularity markers on T-cell surfaces [77]. Nowadays, it is also accepted that functional expansion secondary to high cerebrospinal fluid (CSF) pressure [78]. Consequently, brain lymphatic vasculature provides an important pathway in both fluid and immune cells circulation from CSF to systemic lymph system, suggesting a role in antigen presentation and immune surveillance of the CNS.
It is also believed that simultaneous ionising radiations’ exposure augments the strength of immunotherapy by: 1) a direct and indirect destruction to tumour cells causing cell deaths, 2) an alteration of the cancer stromal microenvironment and 3) activation of CD8 T cells. Radiation induces activation of sequential biological mechanisms and biochemical events, including stimulation of interferon genes pathway and up-regulation of transforming growth factor β, leading to initiation of immune responses [79]. Therefore, co-administration of immunotherapy to block immune checkpoint is beneficial [80]. We will discuss here immune check point inhibitors and the dendritic cell (DC) vaccine as part of immunotherapy strategies in recurrent GBM. At the end, we will highlight genetic syndromes in paediatric GBM patients for whom immunotherapy could be the best choice for them based on recent clinical data.
Immune checkpoint inhibitors (ICIs)
Cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and PD-1 pathways operate at distinct stages of an immune-response, resulting in negative effects on T-cell activity (Figure 4). CTLA-4 pathway is in the early stage of T cell activation, by binding of CD28 molecules on T-cells with B7-1 (CD80) and/or B7-2 (CD86) molecules on the surface of an antigen-presenting cell, mainly in the lymph nodes. On the other hand, PD-1 pathway blocks T cells at a later stage of the immune response through binding to PD-L1 and programmed death ligand 2 (PD-L2), in peripheral tissues [80] as in Figure 4.
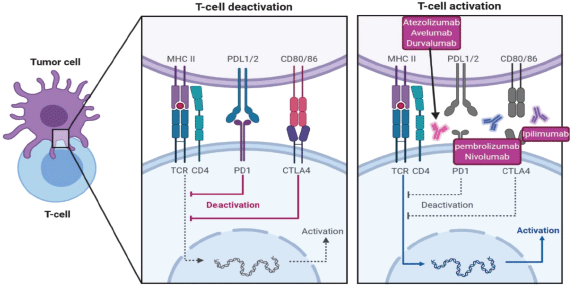
Figure 4. Immunotherapeutic pathways to in-activate T-cell against tumour cells and different immunotherapy to reactivate T-cells, CTLA4 inhibitor ipilimumab, PD1 inhibitor pembrolizumab and nivolumab and PDL-1 inhibitor atezolizumab, avelumab and durvalumab.
Generally, anti-PD-1 drugs have a well-tolerated adverse event profile rather than anti-CTLA-4. It is suggested that a greater toxicity could be related to a better responses. Additional results are needed to decide whether this hypothesis is validated. Despite a different spectrum of adverse events, a safe and effective management of checkpoint inhibitors’ toxicity is mainly based on early sign and symptom recognition.
Overall, objective responses to ICI in rGBM were seen in the following trials shown in Table 2 the most important of them are as follows [81, 82].
In one retrospective analysis which investigated the effect of ipilimumab 3 mg/kg every 3 weeks with bevacizumab found an over-all response rate (ORR) of 31% in patients with rGBM [82]. A phase I trial found that the ORR of nivolumab single agent and nivolumab ipilimumab was 11% and 10%, respectively [83]. Other phase I study used atezolizumab (1,200 mg Q3W) in 16 patients with glioblastoma showed an ORR of 6.0% [84]. Three patients with isocitrate dehydrogenase 1 (IDH1)-mutant tumours had better PFS (5.5 months versus 1.2 months) and slight better OS (16.0 months versus 2.7 months) than patients who had IDH1-wild-type tumours. Some studies looked at the utilisation of immunotherapy as neoadjuvant treatment and one of them found that when pembrolizumab was given before surgery, patients had longer OS than patients who received it as adjuvant therapy only. Neoadjuvant therapy with pembrolizumab was linked to up-regulation of T cell and interferon-γ-related gene expression, and down regulation of genes related to cell cycle progression [85]. Similar changes were observed with neoadjuvant nivolumab in a phase II trial [86].
Ongoing, three important phase III studies investigating the role of nivolumab in newly-diagnosed and recurrent glioblastoma are awaiting the final analysis publication, but the initial results are disappointing, checkmate-143 was the only one included patients with recurrent GBM, the other two trials included primary GBM only. The first trial was a randomised, open-label, phase III CheckMate-143 trial (NCT02017717), nivolumab monotherapy did not show significant change of OS in comparison to bevacizumab in rGBM patients [87]. The second one combined nivolumab with radiotherapy in CheckMate-498 trial (NCT02617589) but also failed to significantly improve OS of patients with newly diagnosed MGMT-unmethylated glioblastoma, in comparison to chemo-radiotherapy with TMZ [88]. Lastly, in MGMT-methylated glioblastoma patients, adding nivolumab to the first-line standard of care chemo-radiotherapy with TMZ in CheckMate-548 (NCT02667587) did not show an effect on PFS which was one of the primary end points, and OS data is still pending [88]. Possible explanation of failure of PD-1 inhibitor nivolumab in checkmate 143 is attributed to impaired interaction between it and PD-1 receptors on patient’s lymphocyte due to either 1) physical barrier by BBB 2) Systemic lymphopenia and 3) Reduced T-cell expression of PD-1 receptors. Poor drug penetration of BBB has been linked to inability of drugs to reach glioma malignant cells. BBB does not allow particles larger than 400–600 Da to pass [89] and nivolumab has calculated molecular mass of 146 kDa [90]. PD-1/programmed death-ligand 1 (PDL-1) axis inhibition occurs outside tumour at lymphoid tissue peripherally then activated coated T-cells enter tumour microenvironment. In the recurrent setting, any activated T cells with PD-1 against the tumour are supposed to have been migrated to tumour site inside CNS where they are inaccessible to mono-clonal antibodies [91] (Figure 5). Another possible reason for failure of checkpoint inhibitors is that patients with GBM have heavily dys-functioning antigen-specific T-cells activation and usually permanently anergic towards tumour’s antigens. This is due to chronic antigen exposure which results in ‘exhausted T-cells’ and over-expression of PDL-1 tumour cells that their function may not be fully regained by PD-1 inhibitor [92, 93]. Single agent with anti-PD-1 is unlikely to reverse all factors that cause T-cell inhibition.
Lastly unlike adult brain GBM, paediatric GBM is linked to genetic syndromes like Li-Fraumeni (Tp53) syndrome and bi-allelic mismatch repair genes syndrome (bMMRD), it is associated with increased incidence of malignancies in the first years of life; most common malignancies include glioblastoma, haematological malignancies and gastrointestinal tract cancers. BMMRD results from homologous germline mutations in one of the following genes (PMS2, MLH1, MSH2 and MSH6). ‘DNA mismatch repair deficient (DMMRD) GBM’ has the highest mutational load in all cancers. Knowing that non-DMMRD cancers with high mutational load like melanoma, lung and bladder malignancies have high response to ICI lead to potential therapeutic opportunity to this subset of patients with GBM. One study did exome sequencing and neoantigen prediction on 37 dMMRD cancers (hyper-mutated) and compared them to adult and paediatric GBM patients, bMMRD GBM had high mutational load compared to sporadic paediatric and adult GBM. Based on this data, two siblings with dMMRD who had recurrent multifocal GBM treated with nivolumab and this treatment resulted in strong radiological response and significant prolongation of clinical response [94].
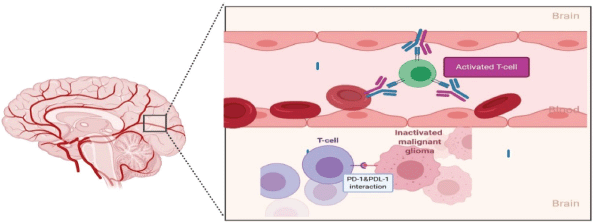
Figure 5. In recurrent disease, efficacy of nivolumab is limited by its inability to cross the BBB and a paucity of functional circulating T-cells with which to interact and form a protective barrier against subsequent possible PD-1/PD-L1 interactions. Exposed to numerous immunosuppressive influences within the glioma microenvironment, including uninhibited PD-1/PD-L1 interactions, T-cells already sequestered within the TME are expected to be heavily dysfunctional and unable to be rescued solely with immune checkpoint inhibition.
DC vaccine
DC is a highly specialised antigen processing and presenting cell which plays a vital role in initiating immune response and useful in immunotherapy as providing a way for cytotoxic T lymphocytes, natural killer cells and cytokines to kill tumour cell directly or indirectly [95]. Several small clinical trials utilised DCs in GBM with conflicting results; some showed no clinical benefit, others showed significant durable response. Recent meta-analysis of six phase II randomised controlled trials [96] included 347 patients with recurrent or primary glioblastoma.
Patients who received DC vaccine had significantly prolonged OS (HR: 0.6995% confidence interval (CI): 0.49 to 0.97, p = 0.03) compared to the control group and a trend towards better PFS was also detected (HR: 0.76, 95% CI: 0.56 to 1.02, p = 0.07).
Moreover, the incidence of side effects was comparable between patients treated with dendritic cell vaccine and control (odds ratio = 1.52, 95% CI: 0.88 to 2.62, p = 0.14; I2= 0%) [96].Some trials reported improvement from 13 month OS in the control group to 15.7–35.9 months OS in those patients who received DCs [95]. So data on this modality of treatment is still immature and results from phase III trials are urgently needed.
Gene therapy in GBM
Gene therapy simply is delivering either tumour suppressor genes to the tumour to regulate its growth by inhibiting oncogenes, or delivering an inactive pro-drug to be activated at the site of the tumour to a lethal compound. GBM gene therapy until now has used different delivery vectors like viral vectors, non-polymeric NPs and polymeric NPs. We will discuss briefly these vectors in the following section [116].
Viral vectors
It is the first and commonly used vectors in GBM like neurotropic retroviruses and adenovirus that possess a specific ability to infect neuron and glial cells like, herpes simplex virus1 (HSV-1) [117, 118]. First trial in retrovirus evaluated HSV thymidine kinase (HSV-TK) which is the suicide gene in combination with ganciclovir (Cytovene) as the pro-drug. HSV-TK will convert the pro-drug ganciclovir to the active form ganciclovir triphosphate which inhibits DNA replication [119]. The results demonstrated limited transfection ability into the tumour [120].
Table 2. Most of the finished and ongoing immunotherapy trials in recurrent GBM.
Toca 511 is another retroviral vector which delivers tumour suppressor gene, cytosine deaminase (CD) and oral pro-drug Toca FC, CD enzyme converts 5-fluorocytosine to active 5-fluorouracil [121]. A phase I trial showed safety of the drug in patients with recurrent GBM with regression of the tumour at the infusion site and now this treatment is being evaluated in phase II/III trials [122].
As regards adenoviral vectors, a phase I trial evaluated adenoviral vector with wild P53 (Ad-p53) transfected into tumour cells showed minimal toxicity but limited ability to penetrate tumour tissue [123, 124]. Another phase Ib trial evaluated adenovirus V-tk with valaciclovir injected post-surgery to tumour bed with concomitant radiation followed by adjuvant TMZ in newly diagnosed patients with GBM [125]. It showed small survival advantage over standard treatment. However, CD3 T cells increased after the new treatment, supporting immune-activation after this therapy. Then a phase II trial with the same treatment versus SOC demonstrated increase in OS from 13.5 months in SOC arm to 17.1 months with the new treatment [126].
Despite many studies of viral vectors in GBM, it resulted only in small survival advantage and the main challenge facing it is the ability to penetrate tumour tissue.
Non-viral vectors
Including polymeric and non-polymeric systems. Few non-polymeric vectors have been studied in GBM including liposomes, gold nanoparticles (NPs) and RNA NP. A transferrin receptor-targeted liposome vector SG-53 encapsulates P53 wild type plasmid DNA can cross BBB targeting glioblastoma cells leading to reduction in MGMT and can induce apoptosis in xenograft mice [127]. So, SGT-53 can increase chemo-sensitivity of TMZ and now under evaluation in phase II trial in combination with TMZ in patients with recurrent GBM [128].
Nu129, a spherical nucleic acid gold NP, contains siRNA targeting B-cell lymphoma 2 (BCL-2) like protein 12 which participates in tumour progression and resistance to apoptosis [129]. Nu129 has proved its ability to cross BBB and increases apoptosis in xenograft mice model and now is under evaluation in phase I trial in patients with recurrent GBM [129].
RNA NPs, for example, tested to deliver anti-miR-21 locked nucleic acid sequences to inhibit miR21 in xenograft GBM models in mice, with significant tumour regression [130]. RNA NP is considered to be a promising treatment although it is still in the pre-clinical phase [131].
Future directions
Based on current evidence and preliminary results of clinical trials assessing new treatment strategies, clustering patients with recurrent GBM according to their molecular profile involved in their disease development will defiantly help optimising decisions in clinical scenarios. This fact can be attributed to the widely varying molecular nature of this disease and its unique micro-environment. The awaited clinical trials results mentioned in this review (Tables 1 and 2) and probably starting phase III trials for the most successful treatment strategies from phase I, II trials will definitely help approving new treatment strategies for these patients.
Also, there is clearly much work to be done to identify novel therapeutic targets and to develop strategies for treating advanced thyroid cancer. Pre-clinical data suggests a number of areas that could be developed in the coming years. For example, GSCs is believed by scientists to be the main cause of relapse as it causes re-growth of the tumour after eradicating the main bulk of it by surgery and chemo radiotherapy [132] (Figure 6). It is important to understand the main pathways that lead to maintenance of GSCs and this area now represents an important scope of research in pre-clinical studies [133]. We mentioned here the most important pathways responsible for GSC maintenance ability, the ‘notch and the Wnt pathways’.
Notch signalling pathway
Notch pathway is important for determination of cell fate, proliferation, maintenance of cell quiescence, migration and regulate neural stem cell differentiation [134]. It starts by activation of γ-secretase by the jagged family ligands which leads to cleavage of notch receptors, then the intracellular notch receptor domain (NICD) translocates to the nucleus and formation of recombination signal binding protein for immunoglobulin kappa J region (RBPJ) and Mastermind-like 1 (MAML) complexes in the nucleus and activation of the hairy and enhancer of split (HES) and HEY genes that maintain multi-potency [135] (Figure 7).
CD 133-positive GSCs over-express genes like ID4 and FABP7 which are notch-pathway activator that leads to enhance infiltration ability of GBM [136]. Due to the above-mentioned function of notch pathway, inactivation of it may be effective for blocking GSCs and limiting sonic hedgehog (SHH/glioma associated oncogene (GLI) signalling pathway which plays an important role in oncogenesis especially neural progenitor regulation. That is why SHH/GLI is an important pathway for self-renewal and tumorigenicity of GSCs in which SHH/GLI pathway is active [137]. Studies found recently that SHH/GLI activity is important for Nanog regulation and expression which is a potent transcription factor and considered as a master regulator of many stem cells [138]. However, P53 decreases GLI activity which leads to Nanog down-regulation. So, P53 loss leads to Nanog up-regulation and maintaining stemness properties. Pre-clinical studies confirmed that this pathway contributes to GSC chemo-resistance and inhibition of SHH could potentiate activity of TMZ [139].
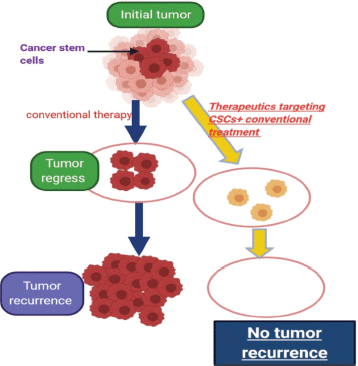
Figure 6. Describing the idea of targeting cancer stem cells in tumors which contain them.
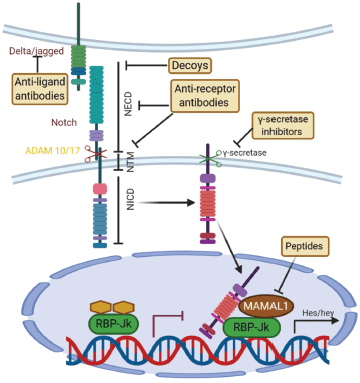
Figure 7. Canonical Notch signaling with points of intervention of current therapies. The interaction between Delta/Jagged-type ligands and Notch receptors leads to S2 cleavage on the extracellular site by “a disintegrin and metalloprotease” 10 (ADAM10) or ADAM17, which is followed by S3 cleavage by the γ-secretase–presenilin complex. The S3 cleavage gives rise to an intracellular Notch fragment (NICD) that translocates into the nucleus, where NICD binds to a protein complex containing recombination signal-binding protein Jκ (RBP-Jκ). This mediates the conversion of RBP-Jκ from a repressor to a transcriptional activator and is followed by the recruitment of the co-activator mastermind-like 1 (MAML1). These events lead to the de-repression of transcription of hairy/enhancer of split (Hes) and Hey. Several stages of the Notch signaling pathway are prone to pharmacological intervention and are labeled in the figure. Gamma-secretase inhibitors and blocking antibodies are already in clinical trials and decoys have been tested in animal models. Peptide inhibitors represent potential future treatment modalities. NECD, Notch extracellular domain; NTM, Notch transmembrane domain.
The Wnt/β-catenin signalling pathway
This pathway is important during CNS development and plays a role in self-renewal, differentiation and neural stem cell development [140]. However, aberrant activation of this pathway in the CNS leads to transformation into brain tumours. There is genetic and epigenetic factors which regulate the association between Wnt pathway and GSC maintenance [141]. Wnt 5A is another member of Wnt family which stimulates endothelial differentiation from GSC then neovascularisation that facilitates tumour growth and invasion [142]. In addition to above-mentioned mechanisms, Wnt signalling promotes (MGMT) expression which leads to TMZ resistance [143]. It is not possible to design a drug that targets Wnt pathway broadly as it is involved in many physiological processes inside brain and in other organs and this will lead to serious side effects. So developing strategies that target Wnt pathway at the tumour level is essential [140].
Conclusion
Recurrent GBM is a fatal disease despite progression in understanding its molecular pathways and trying to target them. EGFR, PDGFR, FGFR and VEGFR inhibitors all showed few advantages although they are amplified or mutated in rGBM. YKL-40 targeting and multi-target of tyrosine receptor kinases may be beneficial. Immunotherapy has many limitations but it is still too early to decide that it is useless, especially DC vaccination which achieved significant advantage in some trials. A future direction towards targeting cancer stem cells pathways like notch and Wnt pathways could be an excellent solution for this disease.
Conflicts of interest
The authors have no conflicts of interest.
Funding statement
The authors have not received any funding for this work.
References
1. Louis DN, Perry A, and Reifenberger G, et al (2016) The 2016 World Health Organization classification of tumors of the central nervous system: a summary Acta Neuropathologica 131(6) 803–820 https://doi.org/10.1007/s00401-016-1545-1 PMID: 27157931
2. Tamimi AF and Juweid M (2017) Epidemiology and outcome of glioblastoma (Exon Publications) pp 143–153. https://doi.org/10.15586/codon.glioblastoma.2017.ch8
3. Stupp R, Taillibert S, and Kanner A, et al (2017) Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: a randomized clinical trial JAMA 318(23) 2306–2316 https://doi.org/10.1001/jama.2017.18718 PMID: 29260225 PMCID: 5820703
4. Keles GE, Lamborn KR, and Chang SM, et al (2004) Volume of residual disease as a predictor of outcome in adult patients with recurrent supratentorial glioblastomas multiforme who are undergoing chemotherapy J Neurosurg 100(1) 41–46 https://doi.org/10.3171/jns.2004.100.1.0041 PMID: 14743910
5. Brandes AA (2005) Adding temozolomide to radiotherapy prolongs survival in people with glioblastoma: abstracted from: Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma N Engl J Med 2005; 352: 987–96 Cancer Treat Rev 31(7) 577–581 https://doi.org/10.1016/j.ctrv.2005.08.004 PMID: 16242242
6. Stupp R, Hegi ME, and Gilbert MR, et al (2007) Chemoradiotherapy in malignant glioma: standard of care and future directions J Clin Oncol 25(26) 4127–4136 https://doi.org/10.1200/JCO.2007.11.8554 PMID: 17827463
7. Qazi MA (2019) A Pre-clinical model of glioblastoma recurrence to identify personalized therapeutic targets 2019
8. Filley AC, Henriquez M, and Dey M (2017) Recurrent glioma clinical trial, CheckMate-143: the game is not over yet Oncotarget 8(53) 91779–91794 https://doi.org/10.18632/oncotarget.21586 PMID: 29207684 PMCID: 5710964
9. Vartanian A, Singh SK, and Agnihotri S, et al (2014) GBM’s multifaceted landscape: highlighting regional and microenvironmental heterogeneity Neuro-oncology 16(9) 1167–1175 https://doi.org/10.1093/neuonc/nou035 PMID: 24642524 PMCID: 4136895
10. Auffinger B, Spencer D, and Pytel P, et al (2015) The role of glioma stem cells in chemotherapy resistance and glioblastoma multiforme recurrence Expert Rev Neurother 15(7) 741–752 https://doi.org/10.1586/14737175.2015.1051968 PMID: 26027432 PMCID: 4830899
11. Network CGAR (2008) Comprehensive genomic characterization defines human glioblastoma genes and core pathways Nature 455(7216) 1061 https://doi.org/10.1038/nature07385
12. Verhaak RG, Hoadley KA, and Purdom E, et al Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1 Cancer Cell 17(1) 98–110 PMID: 20129251 PMCID: 2818769
13. Szerlip NJ, Pedraza A, and Chakravarty D, et al (2012) Intratumoral heterogeneity of receptor tyrosine kinases EGFR and PDGFRA amplification in glioblastoma defines subpopulations with distinct growth factor response Proc Natl Acad Sci 109(8) 3041–3046 https://doi.org/10.1073/pnas.1114033109 PMID: 22323597 PMCID: 3286976
14. Favero F, McGranahan N, and Salm M, et al (2015) Glioblastoma adaptation traced through decline of an IDH1 clonal driver and macro-evolution of a double-minute chromosome Ann Oncol 26(5) 880–887 https://doi.org/10.1093/annonc/mdv127 PMID: 25732040 PMCID: 4405282
15. Nguyen HS, Shabani S, and Awad AJ, et al (2018) Molecular markers of therapy-resistant glioblastoma and potential strategy to combat resistance Int J Mol Sci 19(6) 1765 https://doi.org/10.3390/ijms19061765 PMCID: 6032212
16. Draaisma K, Chatzipli A, and Taphoorn M, et al (2020) Molecular evolution of IDH wild-type glioblastomas treated with standard of care affects survival and design of precision medicine trials: a report from the EORTC 1542 study J Clin Oncol 38(1) 81–99 https://doi.org/10.1200/JCO.19.00367
17. Vivanco I, Robins HI, and Rohle D, et al (2012) Differential sensitivity of glioma-versus lung cancer–specific EGFR mutations to EGFR kinase inhibitors Cancer Discov 2(5) 458–471 https://doi.org/10.1158/2159-8290.CD-11-0284 PMID: 22588883 PMCID: 3354723
18. Lee JC, Vivanco I, and Beroukhim R, et al (2006) Epidermal growth factor receptor activation in glioblastoma through novel missense mutations in the extracellular domain PLoS Med 3(12) e485 https://doi.org/10.1371/journal.pmed.0030485 PMID: 17177598 PMCID: 1702556
19. Nishikawa R, Ji X-D, and Harmon RC, et al (1994) A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity Proc Natl Acad Sci 91(16) 7727–7731 https://doi.org/10.1073/pnas.91.16.7727 PMID: 8052651 PMCID: 44475
20. Weinstein IB (2002) Addiction to oncogenes--the Achilles heal of cancer Science 297(5578) 63–64 https://doi.org/10.1126/science.1073096 PMID: 12098689
21. Yarden Y and Pines G (2012) The ERBB network: at last, cancer therapy meets systems biology Nat Rev Cancer 12(8) 553–563 https://doi.org/10.1038/nrc3309 PMID: 22785351
22. Bianco R, Shin I, and Ritter CA, et al (2003) Loss of PTEN/MMAC1/TEP in EGF receptor-expressing tumor cells counteracts the antitumor action of EGFR tyrosine kinase inhibitors Oncogene 22(18) 2812–2822 https://doi.org/10.1038/sj.onc.1206388 PMID: 12743604
23. Rich JN, Reardon DA, and Peery T, et al (2004) Phase II trial of gefitinib in recurrent glioblastoma J Clin Oncol 22(1) 133–142 https://doi.org/10.1200/JCO.2004.08.110
24. Lassman AB, Rossi MR, and Razier JR, et al (2005) Molecular study of malignant gliomas treated with epidermal growth factor receptor inhibitors: tissue analysis from North American Brain Tumor Consortium Trials 01-03 and 00-01 Clin Cancer Res 11(21) 7841–7850 https://doi.org/10.1158/1078-0432.CCR-05-0421 PMID: 16278407
25. Raizer JJ, Abrey LE, and Lassman AB, et al (2010) A phase II trial of erlotinib in patients with recurrent malignant gliomas and nonprogressive glioblastoma multiforme postradiation therapy Neuro-oncology 12(1) 95–103 https://doi.org/10.1093/neuonc/nop015 PMID: 20150372 PMCID: 2940554
26. de Vries NA, Buckle T, and Zhao J, et al (2012) Restricted brain penetration of the tyrosine kinase inhibitor erlotinib due to the drug transporters P-gp and BCRP Investig New Drugs 30(2) 443–449 https://doi.org/10.1007/s10637-010-9569-1
27. van den Bent MJ, Brandes AA, and Rampling R, et al (2009) Randomized phase II trial of erlotinib versus temozolomide or carmustine in recurrent glioblastoma: EORTC brain tumor group study 26034 J Clin Oncol 27(8) 1268 https://doi.org/10.1200/JCO.2008.17.5984 PMID: 19204207 PMCID: 2667826
28. Camp ER, Summy J, and Bauer TW, et al (2005) Molecular mechanisms of resistance to therapies targeting the epidermal growth factor receptor Clin Cancer Res 11(1) 397–405 PMID: 15671571
29. Reardon DA, Quinn JA, and Vredenburgh JJ, et al (2006) Phase 1 trial of gefitinib plus sirolimus in adults with recurrent malignant glioma Clin Cancer Res 12(3) 860–868 https://doi.org/10.1158/1078-0432.CCR-05-2215 PMID: 16467100
30. Reardon DA, Desjardins A, and Vredenburgh JJ, et al (2010) Phase 2 trial of erlotinib plus sirolimus in adults with recurrent glioblastoma J Neuro-oncol 96(2) 219–230 https://doi.org/10.1007/s11060-009-9950-0
31. Wen PY, Chang SM, and Lamborn KR, et al (2014) Phase I/II study of erlotinib and temsirolimus for patients with recurrent malignant gliomas: North American Brain Tumor Consortium trial 04-02 Neuro-oncology 16(4) 567–578 https://doi.org/10.1093/neuonc/not247 PMID: 24470557 PMCID: 3956354
32. Kreisl TN, Lassman AB, and Mischel PS, et al (2009) A pilot study of everolimus and gefitinib in the treatment of recurrent glioblastoma (GBM) J Neuro-oncol 92(1) 99–105 https://doi.org/10.1007/s11060-008-9741-z
33. Reardon DA, Conrad CA, and Cloughesy T, et al (2012) Phase I study of AEE788, a novel multitarget inhibitor of ErbB-and VEGF-receptor-family tyrosine kinases, in recurrent glioblastoma patients Cancer Chemother Pharmacol 69(6) 1507–1518 https://doi.org/10.1007/s00280-012-1854-6 PMID: 22392572 PMCID: 4351868
34. Reardon DA, Nabors LB, and Mason WP, et al (2015) Phase I/randomized phase II study of afatinib, an irreversible ErbB family blocker, with or without protracted temozolomide in adults with recurrent glioblastoma Neuro Oncol 17(3) 430–439 PMCID: 4483093
35. Sepúlveda-Sánchez JM, Vaz MÁ, and Balañá C, et al (2017) Phase II trial of dacomitinib, a pan-human EGFR tyrosine kinase inhibitor, in recurrent glioblastoma patients with EGFR amplification Neuro-oncology 19(11) 1522–1531 https://doi.org/10.1093/neuonc/nox105 PMID: 28575464 PMCID: 5737732
36. Chen W-S, Hong L, and Wang F, et al (2019) Investigation of dacomitinib on reducing cell necrosis and enhancing cell apoptosis in C6 glioma rat model by MRI Biosci Rep 39(3)
37. Smyth L, Saura C, and Piha-Paul S, et al (2019) 30P Update on the phase II SUMMIT trial: Neratinib fulvestrant for HER2-mutant, HR-positive, metastatic breast cancer Ann Oncol 30(Supplement_3) mdz095. 29 https://doi.org/10.1093/annonc/mdz095.029
38. Viloria-Petit A, Crombet T, and Jothy S, et al (2001) Acquired resistance to the antitumor effect of epidermal growth factor receptor-blocking antibodies in vivo: a role for altered tumor angiogenesis Cancer Res 61(13) 5090–5101 PMID: 11431346
39. Naumov GN, Nilsson MB, and Cascone T, et al (2009) Combined vascular endothelial growth factor receptor and epidermal growth factor receptor (EGFR) blockade inhibits tumor growth in xenograft models of EGFR inhibitor resistance Clin Cancer Res 15(10) 3484–3494 https://doi.org/10.1158/1078-0432.CCR-08-2904 PMID: 19447865 PMCID: 2893040
40. Cohen MH, Shen YL, and Keegan P, et al (2009) FDA drug approval summary: bevacizumab (Avastin) as treatment of recurrent glioblastoma multiforme Oncologist 14(11) https://doi.org/10.1634/theoncologist.2009-0121 PMID: 19897538
41. Sathornsumetee S, Desjardins A, and Vredenburgh JJ, et al (2010) Phase II trial of bevacizumab and erlotinib in patients with recurrent malignant glioma Neuro-oncology 12(12) 1300–1310 https://doi.org/10.1093/neuonc/noq099 PMID: 20716591 PMCID: 3018944
42. Reardon DA, Nabors LB, and Mason WP, et al (2015) Phase I/randomized phase II study of afatinib, an irreversible ErbB family blocker, with or without protracted temozolomide in adults with recurrent glioblastoma Neuro-oncology 17(3) 430–439 PMCID: 4483093
43. Sepúlveda J, Zahonero C, and Hernandez-Lain A, et al (2014) Targeting EGFR in glioblastoma: preclinical testing of dacomitinib J Clin Oncol 32 e13015 https://doi.org/10.1200/jco.2014.32.15_suppl.e13015
44. Touat M, Idbaih A, and Sanson M, et al (2017) Glioblastoma targeted therapy: updated approaches from recent biological insights Ann Oncol 28(7) 1457–1472 https://doi.org/10.1093/annonc/mdx106 PMID: 28863449 PMCID: 5834086
45. Reardon DA, Wen PY, and Mellinghoff IK (2014) Targeted molecular therapies against epidermal growth factor receptor: past experiences and challenges Neuro-Oncology 16(suppl_8) viii7–viii13 https://doi.org/10.1093/neuonc/nou232 PMID: 25342602 PMCID: 4207137
46. Paulsson J, Ehnman M, and Östman A (2014) PDGF receptors in tumor biology: prognostic and predictive potential Fut Oncol 10(9) 1695–1708 https://doi.org/10.2217/fon.14.83
47. Brennan CW, Verhaak RG, and McKenna A, et al Erratum: The somatic genomic landscape of glioblastoma (Cell (2013) 155 (462-477)) Cell 157(3) 753 https://doi.org/10.1016/j.cell.2014.04.004
48. De Witt Hamer PC (2010) Small molecule kinase inhibitors in glioblastoma: a systematic review of clinical studies Neuro-oncology 12(3) 304–316 https://doi.org/10.1093/neuonc/nop068 PMID: 20167819 PMCID: 2940593
49. Dresemann G, Weller M, and Rosenthal MA, et al (2010) Imatinib in combination with hydroxyurea versus hydroxyurea alone as oral therapy in patients with progressive pretreated glioblastoma resistant to standard dose temozolomide J Neuro-oncol 96(3) 393–402 https://doi.org/10.1007/s11060-009-9976-3
50. Frolov A, Evans IM, and Li N, et al (2016) Imatinib and Nilotinib increase glioblastoma cell invasion via Abl-independent stimulation of p130Cas and FAK signalling Sci Rep 6(1) 1–12 https://doi.org/10.1038/srep27378
51. Batchelor TT, Gerstner ER, and Ye X, et al (2017) Feasibility, phase I, and phase II studies of tandutinib, an oral platelet-derived growth factor receptor-β tyrosine kinase inhibitor, in patients with recurrent glioblastoma Neuro-oncology 19(4) 567–575
52. Alexandru O, Sevastre A-S, and Castro J, et al (2019) Platelet-derived growth factor receptor and ionizing radiation in high grade glioma cell lines Int J Mol Sci 20(19) 4663 https://doi.org/10.3390/ijms20194663 PMCID: 6802357
53. Ohgaki H and Kleihues P (2007) Genetic pathways to primary and secondary glioblastoma Am J Pathol 170(5) 1445–1453 https://doi.org/10.2353/ajpath.2007.070011 PMID: 17456751 PMCID: 1854940
54. Ausprunk DH and Folkman J (1977) Migration and proliferation of endothelial cells in preformed and newly formed blood vessels during tumor angiogenesis Microvasc Res 14(1) 53–65 https://doi.org/10.1016/0026-2862(77)90141-8 PMID: 895546
55. Gerstner ER, Eichler AF, and Plotkin SR, et al Phase I trial with biomarker studies of vatalanib (PTK787) in patients with newly diagnosed glioblastoma treated with enzyme inducing anti-epileptic drugs and standard radiation and temozolomide J Neurooncol 103(2) 325–332 PMID: 20821342 PMCID: 4090923
56. Lee EQ, Kuhn J, and Lamborn KR, et al (2012) Phase I/II study of sorafenib in combination with temsirolimus for recurrent glioblastoma or gliosarcoma: North American Brain Tumor Consortium study 05-02 Neuro Oncol 14(12) 1511–1518 https://doi.org/10.1093/neuonc/nos264 PMID: 23099651 PMCID: 3499017
57. Kalpathy-Cramer J, Chandra V, and Da X, et al (2017) Phase II study of tivozanib, an oral VEGFR inhibitor, in patients with recurrent glioblastoma J Neurooncol 131(3) 603–610 https://doi.org/10.1007/s11060-016-2332-5
58. Reardon DA, Groves MD, and Wen PY, et al (2013) A phase I/II trial of pazopanib in combination with lapatinib in adult patients with relapsed malignant glioma Clin Cancer Res 19(4) 900–908 https://doi.org/10.1158/1078-0432.CCR-12-1707 PMID: 23363814
59. Batchelor TT, Mulholland P, and Neyns B, et al (2013) Phase III randomized trial comparing the efficacy of cediranib as monotherapy, and in combination with lomustine, versus lomustine alone in patients with recurrent glioblastoma J Clin Oncol 31(26) 3212–3218 https://doi.org/10.1200/JCO.2012.47.2464 PMID: 23940216 PMCID: 4021043
60. Popescu AM, Alexandru O, and Brindusa C, et al (2015) Targeting the VEGF and PDGF signaling pathway in glioblastoma treatment Int J Clin Exp Pathol 8(7) 7825–7837 PMID: 26339347 PMCID: 4555675
61. Batista KMP, Eulate-Beramendi SA, and Pińa K, et al (2017) Mesenchymal/proangiogenic factor YKL-40 related to glioblastomas and its relationship with the subventricular zone Folia Neuropathol 55(1) 14–22 https://doi.org/10.5114/fn.2017.66709 PMID: 28430288
62. Francescone RA, Scully S, and Faibish M, et al (2011) Role of YKL-40 in the angiogenesis, radioresistance, and progression of glioblastoma J Biol Chem 286(17) 15332–15343 https://doi.org/10.1074/jbc.M110.212514 PMID: 21385870 PMCID: 3083166
63. Lasorella A, Sanson M, and Iavarone A (2017) FGFR-TACC gene fusions in human glioma Neuro Oncol 19(4) 475–483 PMCID: 5464372
64. Jimenez-Pascual A and Siebzehnrubl FA (2019) Fibroblast growth factor receptor functions in glioblastoma. Cells 8(7) https://doi.org/10.3390/cells8070715 PMID: 31337028 PMCID: 6678715
65. Smyth L, Piha-Paul S, and Saura C, et al (2019) Abstract PD3-06: Neratinib fulvestrant for HER2-mutant, HR-positive, metastatic breast cancer: updated results from the phase 2 SUMMIT trial Cancer Res 79(4 Supplement) PD3-06-PD3-.
66. Schramm K, Iskar M, and Statz B, et al (2019) DECIPHER pooled shRNA library screen identifies PP2A and FGFR signaling as potential therapeutic targets for diffuse intrinsic pontine gliomas Neuro Oncol 21(7) 867–877 https://doi.org/10.1093/neuonc/noz057 PMID: 30943283 PMCID: 6620639
67. Alexandru O, Horescu C, and Sevastre A-S, et al (2020) Receptor tyrosine kinase targeting in glioblastoma: performance, limitations and future approaches Contemp Oncol 24(1) 55–66
68. Kreisl TN, McNeill KA, and Sul J, et al (2012) A phase I/II trial of vandetanib for patients with recurrent malignant glioma Neuro Oncol 14(12) 1519–1526 https://doi.org/10.1093/neuonc/nos265 PMID: 23099652 PMCID: 3499018
69. Chheda MG, Wen PY, and Hochberg FH, et al (2015) Vandetanib plus sirolimus in adults with recurrent glioblastoma: results of a phase I and dose expansion cohort study J Neurooncol 121(3) 627–634 https://doi.org/10.1007/s11060-014-1680-2 PMCID: 4324090
70. Loilome W, Joshi A, and Rhys C, et al (2009) Glioblastoma cell growth is suppressed by disruption of Fibroblast Growth Factor pathway signaling J Neuro-oncol 94 359–366 https://doi.org/10.1007/s11060-009-9885-5
71. Naoum GE, Morkos M, and Kim B, et al (2018) Novel targeted therapies and immunotherapy for advanced thyroid cancers Mol Cancer 17(1) 51 https://doi.org/10.1186/s12943-018-0786-0 PMID: 29455653 PMCID: 5817719
72. Kurz SC and Wen PY (2018) Quo vadis—do immunotherapies have a role in glioblastoma? Curr Treat Options Neurol 20(5) 14 https://doi.org/10.1007/s11940-018-0499-0 PMID: 29666934
73. Jackson CM, Choi J, and Lim M (2019) Mechanisms of immunotherapy resistance: lessons from glioblastoma Nat Immunol 20(9) 1100–1109 https://doi.org/10.1038/s41590-019-0433-y PMID: 31358997
74. McGinnis GJ and Raber J (2017) CNS side effects of immune checkpoint inhibitors: preclinical models, genetics and multimodality therapy Immunotherapy 9(11) 929–941 https://doi.org/10.2217/imt-2017-0056
75. Thakar MS, Kearl TJ, and Malarkannan S (2019) Controlling cytokine release syndrome to harness the full potential of CAR-based cellular therapy Front Oncol 9 1529 https://doi.org/10.3389/fonc.2019.01529
76. Rascher G, Fischmann A, and Kröger S, et al (2002) Extracellular matrix and the blood-brain barrier in glioblastoma multiforme: spatial segregation of tenascin and agrin Acta Neuropathol 104(1) 85–91 https://doi.org/10.1007/s00401-002-0524-x PMID: 12070669
77. Yang I, Han SJ, and Kaur G, et al (2010) The role of microglia in central nervous system immunity and glioma immunology J Clin Neurosci 17(1) 6–10 https://doi.org/10.1016/j.jocn.2009.05.006
78. Louveau A, Smirnov I, and Keyes TJ, et al (2015) Structural and functional features of central nervous system lymphatic vessels Nature 523(7560) 337–341 https://doi.org/10.1038/nature14432 PMID: 26030524 PMCID: 4506234
79. Yang B, Wang X, and Ying C, et al (2020) Long Noncoding RNA SNHG16 Facilitates Abdominal Aortic Aneurysm Progression through the miR-106b-5p/STAT3 Feedback Loop J Atheroscler Thromb 52274
80. Pardoll DM (2012) The blockade of immune checkpoints in cancer immunotherapy Nat Rev Cancer 12(4) 252–264 https://doi.org/10.1038/nrc3239 PMID: 22437870 PMCID: 4856023
81. Lukas RV, Rodon J, and Becker K, et al (2018) Clinical activity and safety of atezolizumab in patients with recurrent glioblastoma J Neurooncol 140(2) 317–328 https://doi.org/10.1007/s11060-018-2955-9 PMID: 30073642
82. Carter T, Shaw H, and Cohn-Brown D, et al (2016) Ipilimumab and Bevacizumab in Glioblastoma Clin Oncol (R Coll Radiol) 28(10) 622–626 https://doi.org/10.1016/j.clon.2016.04.042
83. Omuro A, Vlahovic G, and Lim M, et al (2018) Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: results from exploratory phase I cohorts of CheckMate 143 Neuro Oncol 20(5) 674–686 https://doi.org/10.1093/neuonc/nox208 PMCID: 5892140
84. Brahmer JR, Tykodi SS, and Chow LQ, et al (2012) Safety and activity of anti-PD-L1 antibody in patients with advanced cancer N Engl J Med 366(26) 2455–2465 https://doi.org/10.1056/NEJMoa1200694 PMID: 22658128 PMCID: 3563263
85. Cloughesy TF and Mochizuki AY (2019) Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma Nat Med 25(3) 477–486 https://doi.org/10.1038/s41591-018-0337-7 PMID: 30742122 PMCID: 6408961
86. Schalper KA, et al (2019) Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma Nat Med 25(3) 470-6
87. Reardon DA, Brandes AA, and Omuro A, et al (2020) Effect of nivolumab vs bevacizumab in patients with recurrent glioblastoma: the CheckMate 143 phase 3 randomized clinical trial JAMA Oncol 6(7) 1–8 https://doi.org/10.1001/jamaoncol.2020.1024 PMCID: 7243167
88. Sampson JH, Omuro AMP, and Preusser M, et al (2016) A Randomized, Phase 3, Open-Label Study of Nivolumab Versus Temozolomide (TMZ) in Combination with Radiotherapy (RT) in Adult Patients (pts) with Newly Diagnosed, O-6-Methylguanine DNA Methyltransferase (MGMT)-Unmethylated Glioblastoma (GBM): CheckMate-498 (Alexandria: American Society of Clinical Oncology)
89. Miura Y, Takenaka T, and Toh K, et al (2013) Cyclic RGD-linked polymeric micelles for targeted delivery of platinum anticancer drugs to glioblastoma through the blood-brain tumor barrier ACS Nano 7(10) 8583–8592 https://doi.org/10.1021/nn402662d PMID: 24028526
90. Deeks ED (2014) Nivolumab: a review of its use in patients with malignant melanoma Drugs 74(11) 1233–1239 https://doi.org/10.1007/s40265-014-0234-4 PMID: 25022950
91. Reardon DA, Gokhale PC, and Klein SR, et al (2016) Glioblastoma eradication following immune checkpoint blockade in an orthotopic, immunocompetent model Cancer Immunol Res 4(2) 124–135 https://doi.org/10.1158/2326-6066.CIR-15-0151
92. Sznol M and Chen L (2013) Antagonist antibodies to PD-1 and B7-H1 (PD-L1) in the treatment of advanced human cancer (Philadelphia: AACR)
93. Blackburn SD, Shin H, and Freeman GJ, et al (2008) Selective expansion of a subset of exhausted CD8 T cells by αPD-L1 blockade Proc Natl Acad Sci 105(39) 15016–15021 https://doi.org/10.1073/pnas.0801497105 PMID: 18809920 PMCID: 2567485
94. Bouffet E, Larouche V, and Campbell BB, et al (2016) Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency J Clin Oncol 34(19) 2206–2211 https://doi.org/10.1200/JCO.2016.66.6552 PMID: 27001570
95. Anguille S, Smits EL, and Lion E, et al (2014) Clinical use of dendritic cells for cancer therapy Lancet Oncol 15(7) e257–e267 https://doi.org/10.1016/S1470-2045(13)70585-0 PMID: 24872109
96. Lv L, Huang J, and Xi H, et al (2020) Efficacy and safety of dendritic cell vaccines for patients with glioblastoma: a meta-analysis of randomized controlled trials Int Immunopharmacol 83 106336 https://doi.org/10.1016/j.intimp.2020.106336 PMID: 32213460
97. Qin S, Xu L, and Yi M, et al (2019) Novel immune checkpoint targets: moving beyond PD-1 and CTLA-4 Mol Cancer 18(1) 155 https://doi.org/10.1186/s12943-019-1091-2 PMID: 31690319 PMCID: 6833286
98. Majd N, Dasgupta P, and de Groot J (2020) Immunotherapy for Neuro-Oncology Immunotherapy (Berlin: Springer) pp 183–203
99. Reardon DA, Kaley TJ, and Dietrich J, et al (2019) Phase II Study to Evaluate Safety and Efficacy of MEDI4736 (Durvalumab) Radiotherapy in Patients with Newly Diagnosed Unmethylated MGMT Glioblastoma (New Unmeth GBM). (Alexandria: American Society of Clinical Oncology)
100. Peters KB, Archer GE, and Norberg P, et al (2019) Safety of Nivolumab in Combination with Dendritic Cell Vaccines in Recurrent High-Grade Glioma (Alexandria: American Society of Clinical Oncology)
101. Liu EK, Sulman EP, and Wen PY, et al (2020) Novel therapies for glioblastoma Curr Neurol Neurosci Rep 20(7) 19 https://doi.org/10.1007/s11910-020-01042-6 PMID: 32445058
102. Thomas JS, El-Khoueiry AB, and Walters IB, et al (2020) Pharmacodynamic, Safety, and Efficacy Results of a Phase I/II Trial of Intratumoral INT230-6 Alone (IT-01) or in Combination with Pembrolizumab (PEM)(Keynote A10) in Patients with Advanced Solid Tumors (Alexandria: American Society of Clinical Oncology)
103. McGranahan T, Therkelsen KE, and Ahmad S, et al (2019) Current state of immunotherapy for treatment of glioblastoma Curr Treat Options Oncol 20(3) 24 https://doi.org/10.1007/s11864-019-0619-4 PMID: 30790064 PMCID: 6394457
104. Wang X, Guo G, and Guan H, et al (2019) Challenges and potential of PD-1/PD-L1 checkpoint blockade immunotherapy for glioblastoma J Exp Clin Cancer Res 38(1) 87 https://doi.org/10.1186/s13046-019-1085-3 PMID: 30777100 PMCID: 6380009
105. Lim M, Ye X, and Piotrowski AF, et al (2020) Updated Safety Phase I Trial of Anti-LAG-3 Alone and in Combination with Anti-PD-1 in Patients with Recurrent GBM (Alexandria: American Society of Clinical Oncology)
106. Schwarze JK, Duerinck J, and Dufait I, et al (2020) A Phase I Clinical Trial on Intratumoral and Intracavitary Administration of Ipilimumab and Nivolumab in Patients with Recurrent Glioblastoma (Alexandria: American Society of Clinical Oncology)
107. https://clinicaltrials.gov/ct2/show/NCT02311582
108. Pathria P, Louis TL, and Varner JA (2019) Targeting tumor-associated macrophages in cancer Trends Immunol 40(4) 310–327 https://doi.org/10.1016/j.it.2019.02.003 PMID: 30890304
109. Souza-Fonseca-Guimaraes F, Cursons J, and Huntington ND (2019) The emergence of natural killer cells as a major target in cancer immunotherapy Trends Immunol 40(2) 142–158 https://doi.org/10.1016/j.it.2018.12.003 PMID: 30639050
110. Molinero L, Li Y, and Chang C-W, et al (2019) Tumor immune microenvironment and genomic evolution in a patient with metastatic triple negative breast cancer and a complete response to atezolizumab J Immunother Cancer 7(1) 1–9 https://doi.org/10.1186/s40425-019-0740-8
111. Schalper KA, Rodriguez-Ruiz ME, and Diez-Valle R, et al (2019) Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma Nat Med 25(3) 470–476 https://doi.org/10.1038/s41591-018-0339-5 PMID: 30742120
112. Mantica M, Pritchard A, and Lieberman F, et al (2018) Retrospective study of nivolumab for patients with recurrent high grade gliomas J Neurooncol 139(3) 625–631 https://doi.org/10.1007/s11060-018-2907-4 PMID: 29779086
113. Carter T, Shaw H, and Cohn-Brown D, et al (2016) Ipilimumab and bevacizumab in glioblastoma Clin Oncol 28(10) 622–626 https://doi.org/10.1016/j.clon.2016.04.042
114. Blumenthal DT, Yalon M, and Vainer GW, et al (2016) Pembrolizumab: first experience with recurrent primary central nervous system (CNS) tumors J Neuro-oncol 129(3) 453–460 https://doi.org/10.1007/s11060-016-2190-1
115. Chamberlain MC and Kim BT (2017) Nivolumab for patients with recurrent glioblastoma progressing on bevacizumab: a retrospective case series J Neurooncol 133(3) 561–569 https://doi.org/10.1007/s11060-017-2466-0 PMID: 28500559
116. Okura H, Smith CA, and Rutka JT (2014) Gene therapy for malignant glioma Mol Cell Ther 2 21 https://doi.org/10.1186/2052-8426-2-21 PMID: 26056588 PMCID: 4451964
117. Watanabe R and Takase-Yoden S (1995) Gene expression of neurotropic retrovirus in the CNS Prog Brain Res 105 255–262 https://doi.org/10.1016/S0079-6123(08)63302-6 PMID: 7568885
118. Braun E, Zimmerman T, and Hur TB, et al (2006) Neurotropism of herpes simplex virus type 1 in brain organ cultures J Gen Virol 87(Pt 10) 2827–2837 https://doi.org/10.1099/vir.0.81850-0 PMID: 16963740
119. Rainov NG (2000) A phase III clinical evaluation of herpes simplex virus type 1 thymidine kinase and ganciclovir gene therapy as an adjuvant to surgical resection and radiation in adults with previously untreated glioblastoma multiforme Human Gene Ther 11(17) 2389–2401 https://doi.org/10.1089/104303400750038499
120. Ram Z, Culver KW, and Oshiro EM, et al (1997) Therapy of malignant brain tumors by intratumoral implantation of retroviral vector-producing cells Nat Med 3(12) 1354–1361 https://doi.org/10.1038/nm1297-1354 PMID: 9396605
121. Takahashi M, Valdes G, and Hiraoka K, et al (2014) Radiosensitization of gliomas by intracellular generation of 5-fluorouracil potentiates prodrug activator gene therapy with a retroviral replicating vector Cancer Gene Ther 21(10) 405–410 https://doi.org/10.1038/cgt.2014.38 PMID: 25301172 PMCID: 4246057
122. Aghi M, Vogelbaum MA, and Kesari S, et al (2014) AT-02 Intratumoral delivery of the retroviral replicating vector (RRV) TOCA 511 in subjects with recurrent high grade glioma: interim report of phase 1 study (NCT 01156584) Neuro-oncology 16(suppl_5) v8-v. https://doi.org/10.1093/neuonc/nou237.2
123. Lang FF, Bruner JM, and Fuller GN, et al (2003) Phase I trial of adenovirus-mediated p53 gene therapy for recurrent glioma: biological and clinical results J Clin Oncol 21(13) 2508–2518 https://doi.org/10.1200/JCO.2003.21.13.2508 PMID: 12839017
124. Arafat WO, Buchsbaum DJ, and Gómez-Navarro J, et al (2003) An adenovirus encoding proapoptotic Bax synergistically radiosensitizes malignant glioma Int J Radiat Oncol Biol Phys 55(4) 1037–1050 https://doi.org/10.1016/S0360-3016(02)04488-7 PMID: 12605984
125. Chiocca EA, Aguilar LK, and Bell SD, et al (2011) Phase IB study of gene-mediated cytotoxic immunotherapy adjuvant to up-front surgery and intensive timing radiation for malignant glioma J Clin Oncol 29(27) 3611–3619 https://doi.org/10.1200/JCO.2011.35.5222 PMID: 21844505 PMCID: 3179270
126. Wheeler LA, Manzanera AG, and Bell SD, et al (2016) Phase II multicenter study of gene-mediated cytotoxic immunotherapy as adjuvant to surgical resection for newly diagnosed malignant glioma Neuro Oncol 18(8) 1137–1145 https://doi.org/10.1093/neuonc/now002 PMID: 26843484 PMCID: 4933478
127. Kim SS, Rait A, and Kim E, et al (2014) A nanoparticle carrying the p53 gene targets tumors including cancer stem cells, sensitizes glioblastoma to chemotherapy and improves survival ACS Nano 8(6) 5494–5514 https://doi.org/10.1021/nn5014484 PMID: 24811110 PMCID: 4076028
128. Kim SS, Rait A, and Kim E, et al (2015) A tumor-targeting p53 nanodelivery system limits chemoresistance to temozolomide prolonging survival in a mouse model of glioblastoma multiforme Nanomed Nanotechnol Biol Med 11(2) 301–311 https://doi.org/10.1016/j.nano.2014.09.005
129. Jensen SA, Day ES, and Ko CH, et al (2013) Spherical nucleic acid nanoparticle conjugates as an RNAi-based therapy for glioblastoma Sci Transl Med 5(209) 209ra152 https://doi.org/10.1126/scitranslmed.3006839 PMID: 24174328 PMCID: 4017940
130. Lee TJ, Yoo JY, and Shu D, et al (2017) RNA Nanoparticle-Based Targeted Therapy for Glioblastoma through Inhibition of Oncogenic miR-21 Mol Ther 25(7) 1544–1555 https://doi.org/10.1016/j.ymthe.2016.11.016 PMID: 28109960 PMCID: 5498802
131. Shu Y, Pi F, and Sharma A, et al (2014) Stable RNA nanoparticles as potential new generation drugs for cancer therapy Adv Drug Deliv Rev 66 74–89 https://doi.org/10.1016/j.addr.2013.11.006
132. Pointer KB, Clark PA, and Zorniak M, et al (2014) Glioblastoma cancer stem cells: Biomarker and therapeutic advances Neurochem Int 71 1–7 https://doi.org/10.1016/j.neuint.2014.03.005 PMID: 24657832 PMCID: 4119816
133. Sharifzad F, Ghavami S, and Verdi J, et al (2019) Glioblastoma cancer stem cell biology: potential theranostic targets Drug Resist Updat 42 35–45 https://doi.org/10.1016/j.drup.2018.03.003 PMID: 30877905
134. Saito N, Aoki K, and Hirai N, et al (2017) Notch Pathway Activation Predicts Resistance to Bevacizumab Therapy in Glioblastoma (Philadelphia: AACR)
135. Bayin NS, Frenster JD, and Sen R, et al (2017) Notch signaling regulates metabolic heterogeneity in glioblastoma stem cells Oncotarget 8(39) 64932 https://doi.org/10.18632/oncotarget.18117 PMID: 29029402 PMCID: 5630302
136. Kaloshi G, Mokhtari K, and Carpentier C, et al (2007) FABP7 expression in glioblastomas: relation to prognosis, invasion and EGFR status J Neuro-oncol 84(3) 245–248 https://doi.org/10.1007/s11060-007-9377-4
137. Clement V, Sanchez P, and De Tribolet N, et al (2007) HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity Curr Biol 17(2) 165–172 https://doi.org/10.1016/j.cub.2006.11.033 PMID: 17196391 PMCID: 1855204
138. Abou-Antoun TJ, Hale JS, and Lathia JD, et al (2017) Brain cancer stem cells in adults and children: cell biology and therapeutic implications Neurotherapeutics 14(2) 372–384 https://doi.org/10.1007/s13311-017-0524-0 PMID: 28374184 PMCID: 5398995
139. Honorato J, de Faria Lopes GP, and Basile G, et al (2018) Abstract B53: Sonic Hedgehog Inhibition in Glioblastoma Potentializes Temozolomide Effect? (Philadelphia: AACR)
140. Zuccarini M, Giuliani P, Ziberi S, Carluccio M, Iorio PD, Caciagli F, et al. The role of Wnt signal in glioblastoma development and progression: a possible new pharmacological target for the therapy of this tumor. Genes. 2018;9(2):105. https://doi.org/10.3390/genes9020105 PMCID: 5852601
141. Pulvirenti T, Van Der Heijden M, Droms LA, Huse JT, Tabar V, Hall A. Dishevelled 2 signaling promotes self-renewal and tumorigenicity in human gliomas. Cancer research. 2011;71(23):7280-90. https://doi.org/10.1158/0008-5472.CAN-11-1531 PMID: 21990322 PMCID: 3228897
142. Binda E, Visioli A, Giani F, Trivieri N, Palumbo O, Restelli S, et al. Wnt5a drives an invasive phenotype in human glioblastoma stem-like cells. Cancer research. 2017;77(4):996-1007. https://doi.org/10.1158/0008-5472.CAN-16-1693
143. Lee Y, Lee J-K, and Ahn SH, et al (2016) WNT signaling in glioblastoma and therapeutic opportunities Lab Investig 96(2) 137–150. https://doi.org/10.1038/labinvest.2015.140