Repurposing drugs in oncology (ReDO)—cimetidine as an anti-cancer agent
Pan Pantziarka1, 2, Gauthier Bouche1, Lydie Meheus1, Vidula Sukhatme3 and Vikas P Sukhatme3, 4
1Anticancer Fund, Brussels, 1853 Strombeek-Bever, Belgium
2The George Pantziarka TP53 Trust, London KT1 2JP, UK
3GlobalCures, Inc; Newton MA 02459, USA
4Beth Israel Deaconess Medical Centre and Harvard Medical School, Boston, MA 02215, USA
Correspondence to: Pan Pantziarka. Email: pan.pantziarka@anticancerfund.org
Abstract
Cimetidine, the first H2 receptor antagonist in widespread clinical use, has anti-cancer properties that have been elucidated in a broad range of pre-clinical and clinical studies for a number of different cancer types. These data are summarised and discussed in relation to a number of distinct mechanisms of action. Based on the evidence presented, it is proposed that cimetidine would synergise with a range of other drugs, including existing chemotherapeutics, and that further exploration of the potential of cimetidine as an anti-cancer therapeutic is warranted. Furthermore, there is compelling evidence that cimetidine administration during the peri-operative period may provide a survival benefit in some cancers. A number of possible combinations with other drugs are discussed in the supplementary material accompanying this paper.
Keywords: drug repurposing, cimetidine, immunostimulant, ReDO project
Copyright: © the authors; licensee ecancermedicalscience. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Current usage
Introduction
Cimetidine (CIM) is a histamine H2-receptor antagonist (H2RA), in the same class as ranitidine, famotidine, and nizatidine. By blocking the action of histamine on gastric parietal cells, the H2RAs are able to reduce the production of gastric acid, and indeed, CIM, the first drug in this class, was developed as a treatment for dyspepsia. In addition to dyspepsia, it is also used clinically for the treatment of peptic ulcers and for gastroesophageal reflux disease (GERD). The original trade name was Tagamet (GlaxoSmithKline), but the drug is now widely available as a generic. It is available as an over-the-counter drug in some countries, including the USA.
Dosage
Oral CIM is available in tablet form and as a liquid suspension; it can also be used intravenously. Tablets are commonly available as 200 mg, 400 mg, and 800 mg doses. For the treatment of gastric or duodenal ulcer, the adult dosage varies between 800 mg and 1600 mg a day, either as single or divided doses throughout the day, for a period of 4–8 weeks. For reflux oesophagitis, the dose is 400 mg four times a day, for a period of 4–8 weeks. CIM is also used for maintenance therapy of gastric ulcers and short bowel syndrome at a daily dose of 400 mg, with a long-term treatment extending to greater than 10 years in some cases [1].
Toxicity
CIM has low toxicity, with the most common side effects being headache, dizziness, diarrhoea, and rash. Rare side effects include gynaecomastia, reversible impotence (particularly reported in patients receiving very high doses, for example, in the treatment of Zollinger-Ellison Syndrome) and, very rarely, galactorrhoea. Rarely, CIM has also been associated with reversible leukopenia and thrombocytopenia, effects that may be particularly important to watch for in cancer patients who may be undergoing chemotherapy [2]. CIM is contraindicated during pregnancy. In children over the age of one, oral CIM may be used at a dose of 25–30 mg/kg body weight per day in divided doses. For children below the age of one, a dose of 20 mg/kg body weight per day in divided doses has been used [3].
Pharmacokinetics
The bioavailability of CIM is 60–75%, with an elimination half-life of about 2–3 hours. Elimination is mainly via the kidneys, with excretion of the unchanged drug between 60% and 40% depending on the dose and method of administration [4]. Plasma concentrations peak at around one hour if taken without food, or after 2 hours with food. When taken without food, there is a second spike in plasma concentration after around 3 hours. Peak plasma concentration is barely affected by food, with values of 1.18 µg/ml and 1.09 µg/ml, respectively, after an oral dose of 200 mg [5]. Plasma concentrations during continuous treatment with 1.0 g/day were above 1.0 µg/ml for 9 out of 24 hours [4].
CIM is an inhibitor of cytochrome P450, through multiple enzymes (including CYP1A2, CYP2D6, and CYP3A, CYP3A3/A4, CYP2C9, and CYP2C18), which may have a significant impact on the metabolism of a wide range of drugs [6, 7]. In terms of chemotherapy, there is evidence, for example, that concomitant use of CIM and epirubicin can raise the area under the curve (AUC) of both the parent compound and the main metabolites significantly in patients with breast cancer [8]. While this suggests caution in clinical use of CIM for cancer patients, there is also a potential for therapeutic benefit if this effect is deliberately utilised to increase the plasma level or delay clearance of drugs being used for anti-cancer purposes, such as the benzimidazole anti-helminthic mebendazole [9].
Other relevant interactions
CIM has been investigated as an agent to reduce side effects of some current treatments. Examples include the use of oral CIM (800 mg daily) to reduce side effects radiotherapy of the para-aortic lymph nodes in the treatment of cervical cancer [10], and in the prevention of vinorelbine-induced phlebitis using intravenous CIM [11]. Nephrotoxicity is a serious side effect of cisplatin, an adverse effect mediated by the organic cation transporter 2 (OCT2) regulated uptake of the drug in proximal tubules. CIM is a potent inhibitor of OCT2, and in vitro investigations showed that it did not alter the uptake or cytotoxicity of cisplatin in an ovarian cancer cell line (IGROV-1) expressing high levels of OCT2. Subsequent tests in a mouse model showed no diminution of the anti-tumour effect of cisplatin. In patients with head and neck cancer, concurrent CIM (800 mg twice a day) did not alter the exposure to unbound cisplatin, a proxy measure of anti-tumour activity [12].
Pre-clinical evidence in cancer—in vitro and in vivo
Early interest in the potential anti-cancer action of CIM was aroused by investigations into the relationship between histamine levels and cancer, particularly by the finding that histamine levels were increased in distant non-cancer tissues in tumour-bearing mice [13, 14] and decreased in plasma, a result confirmed in humans [15]. An early investigation suggested that a histamine H2 receptor agonist stimulated cellular proliferation in dimethylhydrazine-induced colonic carcinoma in rats, an effect reversed by CIM and another H2RA (metiamide), and that an H1 receptor antagonist had no effect [16]. Initial results also suggested that treatment with metiamide could slow tumour growth and increase survival in sarcoma-bearing mice [17].
Subsequent in vivo results were also obtained with CIM in a number of rat and mouse models by different research groups, with attention focused in particular on immunomodulatory mechanisms [18–20]. Other early in vivo results indicated that CIM could enhance the cytotoxic effect of cyclophosphamide in male DBA2 mice injected with P-388 leukaemia cells, significantly increasing survival time [21]. However, there were issues with study replication, and it was reported by Hannant et al that in vivo results from other groups could not be reproduced [22]. Similarly, the potentiation of the cytotoxic effect of cyclophosphamide could not be replicated with a different mouse model, although a potentiation of the anti-tumour effect of razoxane was reported [23].
One possible explanation for these mixed results was suggested by the results in immunocompetent and immunosuppressed DBA2 mice, which showed that CIM reversed the accelerated growth of implanted tumours in immunosuppressed mice known to have higher levels of suppressor cell activity but had no effect on normal mice (though in one of three experiments CIM treatment increased tumour growth in normal mice, a result possibly due to atypically slow tumour growth in this experiment) [24].
The effect of CIM, often in combination with other agents, particularly immunomodulators such as interferon or IL2, has continued to be explored in numerous in vivo studies in a range of cancer types, including melanoma [25], ovarian cancer [26], colorectal cancer [27], gastric tumours [28], pancreatic cancer [29], lung cancer [30], and gliomas [31] in the years since the early 1980s. It is beyond the scope of this paper to fully summarise this wide range of studies, only a few of which have been referenced here, particularly as there is a comparable range of clinical work being carried out and which is summarised in the next section. However, it should be noted that it is the clinical evidence that is of primary interest from the point of view of drug repurposing [32].
Human data in cancer
One of the earliest references to an anti-cancer effect of CIM comes from a report of two cases published in Lancet in 1979 [33]. In both cases, patients with metastatic disease had displayed some tumour regression following treatment with CIM. This, and the subsequent flurry of animal results that focused primarily on the putative immunomodulatory role of CIM, initiated many clinical studies in the use of CIM in oncology. A full survey is beyond the scope of this paper, but the main results are summarised below, listed for those cancers for which there is the highest level of clinical evidence.
Colorectal cancer
Based on earlier in vitro and in vivo results [27], Adams and coworkers investigated the use of perioperative CIM in patients undergoing surgical resection of colorectal cancer. Control patients showed significant falls in lymphocyte proliferation and cell-mediated immunity. In contrast, patients treated with oral CIM at a dose of 400 mg twice a day for a minimum of 5 preoperative days, then intravenously for 2 post-operative days, showed no significant falls in either lymphocyte proliferation or cell-mediated immunity, indicating that CIM helped reduce post-operative immunosuppression following resection [34]. There was some indication that this difference could provide some clinical benefit in a follow-up that looked at survival at 3 years in two subsequent reports, which showed that with a median follow-up at 30 months, the calculated 3-year survival was 93% for CIM-treated patients and 59% for controls [35, 36].
In a randomised, blinded trial, Svendsen et al treated 192 patients with oral CIM, at a dose of 400 mg twice a day, following surgery (resection or exploratory) for colon (123 patients) or rectal (69 patients) cancer [37]. CIM treatment commenced in the first 3 weeks following surgery and continued for 2 years. The primary end point was cancer-specific mortality. In patients treated with curative intent (148 patients), there was no difference in this end point between the treatment and control arms. However, on stratification, the curatively treated patients with Dukes C disease tended towards lower cancer-specific mortality, though this did not reach statistical significance (29% reduction, 90% confidence interval 2–57%, p = 0.11 log-rank test). There were no differences between groups in the non-curatively treated patients.
In another double-blinded trial of preoperative CIM in Australia, 112 colorectal cancer patients were randomised to low-dose (400 mg twice a day), high-dose (800 mg twice a day), or placebo arms [38]. Treatment was given for five days prior to surgical excision. Kaplan-Meier survival analysis showed there were no significant differences between treatment groups, although there was a trend towards survival advantage in the high-dose CIM (800 mg) group (p = 0.20, log-rank test). However, stratification by replication error (RER) positive or negative tumours showed a statistically significant difference between the high-dose CIM group and the placebo group (p = 0.04, log-rank test) for patients with RER negative tumours.
An unblinded randomised multi-centre trial in Japan reported on long-term survival of colorectal cancer patients treated post-operatively with oral CIM, at a dose of 800 mg daily [39]. A total of 72 patients with colorectal cancer and a primary tumour of T2 or T3 were enrolled, after exclusion of patients who had previously been treated with chemotherapy, radiotherapy, or immunotherapy or who had multiple cancers or severe complications. Of these 72 patients, two did not undergo curative resection, three did not receive adequate drug administration, and three whose disease stage was considered inappropriate for the trial were further considered ineligible and were excluded from the analysis. The remaining 64 patients were randomly allocated, and there were none lost to follow-up. All patients underwent curative resection and then received 200 mg per day of oral 5-U for one year. The treatment group of 34 patients additionally received 800 mg per day of CIM for the 1-year treatment period. In both groups, treatment started 2 weeks post-surgery. Mean follow-up was 10.7 years and showed that the 10-year survival rate of the treatment group was 84.6%, whereas that of a control group was 49.8% (p < 0.0001).
A trial of oral CIM during the perioperative period in 49 patients suffering from gastrointestinal tumours investigated the effect on the immune response, including 19 patients suffering from colorectal cancer [40]. Patients were randomised to perioperative CIM (24 patients) or control (25 patients), with an equal number of colorectal cancer patients (19) in each arm. The patients in the treatment group received 400 mg of oral CIM three times a day for 7 days prior and up to the day of surgery, and then intravenous CIM at 600 mg twice a day during and post-surgery for 10 days. The control arm received standard of care without CIM. The primary end points were measurement of immune status, including peripheral blood lymphocytes, natural killer (NK) cells and tumour-infiltrating lymphocytes (TIL). No clinical outcomes were assessed. In comparison with blood counts from healthy individuals, both treatment and control arms showed decline in the proportion of total T cells, T helper cells, and NK cells. These changes were reversed in the patients in the CIM arm, who showed significantly higher counts on the 10th post-operative day than controls. Also significant was the difference in the number of patients showing increases in TIL response: 68% (17/25) of the patients in the treatment group had significant TIL responses, and only 25% (6/24) of the cases had discernible TIL responses (p < 0.01).
A 2012 Cochrane Review of H2RAs as adjuvant treatments for resected colorectal cancer pooled data from six randomised clinical trials, five of which utilised CIM and one used ranitidine. The review found a trend towards improved survival when H2RAs were utilised as adjuvant therapy in patients having curative-intent surgery for colorectal cancer (HR 0.70; 95% CI 0.48-1.03, P = 0.07). However, analysis of the five CIM trials (with pooled data for 421 patients) found a statistically significant improvement in overall survival (HR 0.53; 95% CI 0.32 to 0.87) [41]. Overall the authors concluded that: cimetidine appears to confer a survival benefit when given as an adjunct to curative surgical resection of colorectal cancers.
Melanoma
The earliest clinical evidence of an effect of the CIM on melanoma was in a series of three cases of recurrent malignant melanoma being treated with coumarin (at a dose of 100 mg per day). Oral CIM was started at a dose of 1000 mg per day when these patients were no longer responding to coumarin treatment. In these cases there was rapid regression of multiple lesions and a corresponding and long-lasting improvement in physical condition. In one further case, recurrent disease was treated with a lower dose of coumarin (25 mg) and CIM (1000 mg), but the disease progressed rapidly, and the patient died shortly after. This patient had not previously been treated with coumarin [42].
A similar pattern was recorded in a series of six melanoma patients, five of whom had disseminated disease, treated with human leukocyte interferon-alpha, with little evidence of effect. The addition of CIM after a period of 6–8 weeks led to a remarkable change, with complete remissions seen in two patients, a partial remission in one, and disease stabilisation in another [43]. The same authors subsequently reported on 20 patients who had also been treated with interferon-alpha with no objective responses; the subsequent introduction of CIM led to six objective responses, including five complete regressions and one extensive partial regression, and three cases of prolonged disease stabilisation [44].
At that time, the standard of care treatment for metastatic melanoma was treatment with dacarbazine or nitrosoureas, with an objective response rate of around 15% and median overall survival of 4 months [45].
A series of seven Phase II studies, in 191 patients, on the use of recombinant interferon-alpha 2a (rIFN-2a), alone and in combination with other agents, for the treatment of disseminated malignant melanoma was carried out by Creagan et al. One of these studies included oral CIM, at a dose of 1200 mg per day (300 mg QID), as an immunostimulant [46]. The response rate for these diverse studies ranged from 0% to 23%, with an aggregate response rate of 14% (27 of 191 patients), and median survival time was 6 months [47]. The best responses, at 23%, were for rIFN-2a monotherapy or rIFN-2a with CIM, suggesting no additional benefit for CIM, although there was a lower level of toxicity in the CIM group.
A later Phase II study, by a different group, looked at the combination of interferon-alpha 2b, IL-2, and cisplatin in metastatic melanoma in a group of 87 patients, with and without oral CIM. An overall response rate of 27% was achieved in the 82 patients evaluable for response, with median response duration of 7 months and median survival of 10.1 months. There were no significant differences between the CIM and non-CIM arms of the study [48].
It is possible that the difference in outcomes between the earlier trials and the more disappointing later trials may be related to the different formulations of interferon used. The form of interferon used by Flodgren et al was human leukocyte derived (HuIFN-alpha (Le)) [43, 44], whereas the work of Creagan et al [47] and Schmidt et al [48] used recombinant interferon 2 alpha. Human-derived interferons contain a wider range of alpha interferon subtypes than recombinant interferons, which are normally restricted to alpha 2a or 2b, and there is some anecdotal evidence that recombinant interferons may not be as effective in stopping tumour development [49]. It may be hypothesised that CIM interacts with some additional alpha interferon subtypes to potentiate the effect and to improve response to treatment, as shown in the earlier trials.
Investigation of CIM as a monotherapy in metastatic melanoma was also investigated in two small Phase II trials, by different groups. In the first trial, 19 previously untreated patients were treated with oral CIM at 1200 mg (300 mg QID). Objective responses were observed in three (19%) patients, including one long-lasting (16 months) complete response, one near-complete response (21 months), and one partial response (7 months). Overall median time to progression was 1.4 months, and overall survival was 6 months [45]. A later trial of CIM monotherapy involved 15 treatment-naïve patients who were treated with high-dose CIM (600 mg QID), although three patients experienced stable disease for 2–4 months. There were no objective responses recorded, and median survival time was 5.3 months, suggesting little significant activity as a monotherapy in this group of patients, although toxicity was negligible even at this high dose [50].
Gastric
Early concerns were raised that the treatment of duodenal ulcers with CIM might alleviate the symptoms of gastric carcinoma, thereby masking disease progression [51], or that CIM itself may be carcinogenic and increase the risk of gastric cancers [52]. However, long-term post-marketing surveillance has shown no such association [53].
A report from Denmark assessed overall survival of gastric cancer patients treated with oral CIM at 800 mg per day (400 mg BID) for 2 years. In this double-blinded study, 181 patients were randomised to CIM or placebo immediately after surgery or the decision not to operate. Median survival in the CIM group was 450 days and 316 days in the placebo group, a statistically significant result (p = 0.02, log-rank test). Relative survival rates (CIM/placebo) were 45%/28% at 1 year, 22%/13% at 2 years, 13%/7% at 3 years, 9%/3% at 4 years, and 2%/0% at 5 years [54].
However, a larger randomised, double-blinded trial in the UK, involving 442 patients, did not find a positive effect of oral CIM [55]. Patients were randomised to either low-dose (400 mg, BID) or high-dose (800 mg, BID) CIM or placebo until tumour progression, recurrence, or death. The median survival for patients receiving CIM was 13 months (95% confidence interval, 9–16 months) and 11 months for the placebo arm (95% confidence interval, 9–14 months), a result that did not reach statistical significance. Within the CIM arms, median survival for the high-dose group was 13 months (95% CI 7–20 months), and 13 months (95% CI 8–18 months) for the low-dose. The 5-year survival was 21% for those randomised to CIM compared with 18% in the placebo arm, again a result that did not achieve statistical significance.
Renal cell carcinoma
The earliest clinical evidence that CIM might have some effect on renal cell carcinoma (RCC) was from a small trial that looked at the combination of CIM and coumarin in 45 patients suffering from metastatic RCC [56]. Patients were treated with coumarin, 100 mg orally daily; on day 15 of treatment, oral CIM was started at a dose of 1200 mg (300 mg QID), and treatment with both drugs was continued until disease progression. Of 42 evaluable patients, there were three complete responses (CR) and eleven partial responses (PR), giving an objective response rate of 33.3%. Twelve patients exhibited stable disease (SD). The median duration of the PR group was 5 months (in the range 4–21 months), while the median duration of the SD group was 7.3 months (in the range 4–16.5 months). There were no reported toxicities with the treatment. Subgroup analysis showed that there were no objective responses in the 14 patients who had not undergone nephrectomy, whereas the fourteen objective responses occurred in the 31 patients who had undergone nephrectomy.
A number of subsequent studies were unable to reproduce this encouraging result. A similar protocol was used in a three-centre Phase II trial that enrolled 31 patients, the majority of whom (84%) had been nephrectomised [57]. Whereas the original study used CIM at 300 mg four times a day, this trial used a dose of 400 mg three times a day, in all other respects the protocol was the same. Of the 31 patients treated, only two (6.5%) showed a PR of 63 weeks and 73 weeks. Both patients experienced regression of pulmonary metastases. Five patients experienced SD (in the range 28–45 weeks). Similarly, another small study used a protocol identical to the original to treat 25 patients, 21 of whom had been nephrectomised. Here there were no objective responses recorded, although five patients experienced SD for more than 3 months. One possible explanation for the disparity may be explained by the better performance status and lower tumour burden of the patients in the original study [58].
CIM has also been investigated in combination with human lymphoblastoid interferon-alpha (LIFN-a) in RCC. A total of 37 patients with advanced RCC were treated between 1982 and 1995 in Japan, of whom 21 patients had metastatic disease at presentation, and 15 had recurrence after nephrectomy. LIFN-a was administered intramuscularly at 5 million units (MU) daily for 5 to 7 days a week for at least 8 weeks, and CIM was administered orally at 200 mg QID [59]. Treatment resulted in an objective response rate of 41%, with a CR in seven patients and a PR in eight. Additionally, 12 patients exhibited SD. Patients with lung metastases showed the best response to therapy. The 5-year survival rates for patients with and without response and overall were 74%, 20%, and 41%, respectively. Histopathologically, high-grade tumours had a better response to combined therapy than did low-grade tumours.
A subsequent Phase III trial by the same group compared treatment of LIFN-a alone with LIFN-a and CIM, with 36 patients recruited to LIFN-a alone and 35 patients to combined LIFN-a and CIM. Intention-to-treat analysis showed one CR, four patients with PR, 16 with SD, and 12 with progressive disease (PD) among the 36, with an overall response rate of 13.9%. Of the 35 patients in the LIFN-a and CIM arm, there were two cases of CR, 8 patients with PR, 13 with SD and 11 with PD, yielding a response rate of 28.6% (P = 0.13). Time to progression ranged from 9 to 845 days (median 112 days) in the LIFN-a group, and from 31 to 1,568 days (median 125 days) in the LIFN-a plus CIM group (P = 0.87) [60]. While tending towards improved response, the authors concluded that the addition of CIM to LIFN-a did not significantly improve the response rate compared to LIFN-a alone.
Despite this conclusion, interest in the combination of interferon and CIM for advanced RCC continues with the addition of other agents. For example, the combination of LIFN-a, CIM, the COX-2 inhibitor meloxicam and the angiotensin II receptor antagonist candesartan was investigated in the Phase II (I-CCA) trial involving 51 patients, of whom 37 (73%) had received prior nephrectomy [61]. Patients received 3–6 MU of LIFN-a, 400 mg CIM BID, 10 mg meloxicam daily and 4 mg candesartan daily. Initially the angiotensin converting enzyme (ACE) inhibitor perindopril erbumine was used, but as this caused a persistent cough in some patients, it was replaced with candesartan. CR was observed in four patients (8%) and PR in seven (14%), giving an overall response rate of 22%. None of the four CR patients relapsed during the 16–81 month follow-up. Of the remaining patients, 24 patients (45%) had SD for at least 6 months, yielding a clinical benefit in 67% of patients, with no grade 3/4 toxicities observed. The median progression-free survival and overall survival were 12 and 30 months, respectively. These results were sufficient for the authors of the study to conclude that the therapy was a potential first-line treatment for advanced RCC that needed to be confirmed in a large international Phase III trial.
The use of high-dose CIM as a single agent in metastatic RCC has also been the subject of a Phase II clinical trial involving 42 patients in the United States. Patients, of whom 38 were evaluable, were treated with 600 mg CIM QID. Two patients showed CR, one of 26 months and one of 33 months, yielding an objective response rate of 5.3%. There were no cases of PR and four cases of SD (duration in the range 3–9 months). Both patients with CR had experienced prior nephrectomy [62]. At this relatively high dose, toxicity was minimal, with one case of mild leukopenia reported.
Other cancers
In addition to the clinical investigations in colorectal and gastric cancer, melanoma, and RCC, there have been a few clinical studies in other cancer types. An investigation of a possible correlation between preoperative CIM and measures of tumour cell proliferation (Ki-67 staining) in breast cancer found no association [63].
In pancreatic cancer, there is a recently published case report of activity of the anti-angiogenic agent TL-118, which includes CIM as one of four drugs that make up the combination agent [64]. In this case report, a 75-year old woman with radiologically confirmed inoperable pancreatic cancer has been treated with TL-118 and gemcitabine and has shown a long-lasting (16 months) progression-free survival. Treatment interruption correlated with an increase of tumour marker CA 19-9, and resumption of treatment reduced levels of this marker.
In metastatic prostate cancer, an early trial looked at the combination of CIM and coumarin, using the same protocol as that for melanoma and RCC [65]. While no objective responses were reported in the fourteen patients in the trial, three patients experienced significant reduction in pain from bone metastases and decreased analgesic use that persisted until disease progression at 3, 5.5 , and 9 months.
A small trial in 28 advanced serous ovarian carcinoma patients found that standard platinum-based chemotherapies augmented with CIM, at a dose of 800 mg/day, commencing 2 weeks before surgery and continuing synchronously with chemotherapy, showed statistically significant improvements in overall survival compared to platinum-based chemotherapy alone [66].
The use of oral CIM in the treatment of Kaposi’s Sarcoma in patients with AIDS was investigated in eight patients with progressive disease (PD) [67]. CIM was given orally at a dose of 300 mg QID, rising to 600 mg QID if there was no response within one month. Of the eight patients evaluated for response, one showed a complete remission of 7 months, one patient had a partial response of 8 months and one showed a mixed response of initial regression followed by PD. The other five patients all showed PD. No patients reported toxicity, and several reported symptomatic improvements.
Clinical trials
TL-118 is a novel drug combination produced by Tiltan Pharma Ltd, Israel. Designed as a multi-targeted anti-angiogenic agent, the four drugs that make up the combination are: CIM, low-dose cyclophosphamide, diclofenac, and sulfasalazine. TL-118 is formulated as an oral suspension and is designed to be taken by patients at home rather than administered in a clinical setting. Currently, there are three Phase II clinical trials of TL-118:
NCT01509911 is an international multi-centre trial in metastatic pancreatic cancer for patients starting gemcitabine treatment. The primary outcome is the disease control rate after 16 weeks of treatment.
NCT01659502 is a single centre study in pancreatic cancer. The primary outcome is a clinical benefit measurement (a composite score based on pain, performance status, and weight) in a 2-year time frame.
NCT00684970 is a multi-centre, single-country trial designated as a Phase IIB trial for metastatic castration-resistant prostate cancer. The primary end point is progression-free survival from 24 weeks after commencement of treatment up to 3 years. Secondary end points include overall survival, time to prostate-specific antigen (PSA) progression, PSA response, and pain response in evaluable patients.
A randomised, double-blinded Phase II trial in Australia and New Zealand (ACTRN12609000769280) is investigating the perioperative use of CIM in patients with colorectal cancer treated with curative resection. The dose is 800 mg tablets twice daily for 5 weeks, starting a week before surgery. The primary outcome is 2-year disease-free survival, with additional subgroup analysis of patients with positive tumour staining for sialyl Lewis antigens. Secondary end points include longer-term disease-free survival, overall survival, and duration of postoperative inflammatory cytokine elevation, assessed as the time that plasma concentrations of each cytokine (TNF, IL-1B, IL-6, IL-8) are elevated above pre-treatment baseline. Recruitment to the trial is complete, and 45% of the cohort have rectal cancer [68].
Mechanism of action
The anti-tumour action of CIM has been shown to be due to four distinct mechanisms:
• Anti-proliferative action on cancer cells
• Immunomodulatory effects
• Effects on cell adhesion
• Anti-angiogenic action
Cancer cell proliferation
It has been shown, in vitro and in vivo, that multiple tumour types express the histamine-synthesising enzyme, L-histidine decarboxylase (HDC) and that tumours can secrete high levels of histamine in a paracrine and/or autocrine fashion. Histamine is highly pleiotropic, with multiple functions involving inflammatory immune response, gastric acid secretion, and action as a neurotransmitter. These diverse physiological actions are mediated by four histamine receptors, of which H2 and H4 are implicated in cancer cell proliferation, invasion, and angiogenesis [69, 70].
A direct effect on cancer cell proliferation has been shown in two xenograft experiments in which exogenous histamine increased tumour growth in C170 and LIM2412 human colorectal cell lines implanted in Balb/c nu/nu mice, an effect that was reversed by oral CIM but not by the H1RA diphenhydramine [27]. Similar results have been shown in gastric cancer, with locally applied histamine increasing the proliferation of implanted MKN45G xenografts in nude mice, an effect abrogated by CIM [71]. In vitro analysis in both colorectal and gastric cancer cell lines showed that the dose-dependent increase in cellular proliferation induced by histamine was associated with an accumulation of cyclic adenosine monophosphate (cAMP) [27, 71].
However, there is also some evidence to suggest that some anti-proliferative effects of CIM may not be entirely related to generic activity as an H2RA. A comparison of the anti-proliferative effect of different H2RAs in gastric cancer cell lines showed that CIM significantly reversed histamine-stimulated proliferation in a dose-dependent manner, ranitidine had a lesser effect and famotidine showed no effect [72]. This suggests either that there is something specific about the binding of CIM to the H2 receptor or else there are additional off-target effects of CIM action. For example, there is also some evidence CIM can cause apoptosis in the Caco-2 human colorectal cancer cell line independent of its action as an H2RA [73]. Similarly, while CIM synergised with a novel phospho-valproic acid to inhibit pancreatic tumour growth in mouse models, another H2RA, ranitidine, showed no such activity [74].
Immunomodulation
Histamine has multiple effects on both innate and adaptive immune responses, mediated by the four histamine receptors (H1–H4). In relation to cancer, histamine is associated with an immunosuppressive tumour microenvironment, including an increase in CD4 CD25 regulatory T cell (Treg) activity, reduced antigen-presenting activity of dendritic cells (DC), reduced NK-cell activity and increased myeloid-derived suppressor cell (MDSC) activity [75–77].
In particular, histamine binding to the H2 receptor is associated with suppression of IL-12 and stimulation of IL-10 secretion and is implicated with a shift in Th1/Th2 balance toward Th2-dominance of the immune response. This effect was reversed by CIM in human PBMC [78]. Similarly, in an HDC knock-out mouse model, animals inoculated subcutaneously with the LM2 murine breast cancer cell line showed slower tumour growth than in HDC wild-type mice. The knock-out mice, lacking endogenous histamine, showed a predominance of Th1 cytokines and a lower level of Foxp3 (associated with CD4 CD25 Tregs) expression compared to wild-type tumour-bearing mice [79].
In addition to Treg cells, the other key drivers of the immunosuppressive tumour microenvironment are MDSC cells, involved in extensive cross-talk with Tregs in promoting T-cell dysfunction and in skewing the immune response towards Th2 [80]. MDSCs express H1–H3 receptors, and there is in vitro and in vivo evidence that blockade of H1 (using the H1RA cetirizine) or H2 (using CIM), can reverse the immunosuppressive action of these cells [77]. For example, the addition of CIM reduced the tumour burden in a B16 melanoma mouse model [77] and in a mouse model of 3LL lung tumour [30].
CIM has also been shown to increase the in vitro antigen-presenting activity of monocyte-derived DC, in advanced colorectal cancer patients compared to controls [81]. An increase in NK activity compared to non-CIM-treated controls has also been noted in cardiopulmonary bypass surgery [82].
Additionally, perioperative CIM has been shown to reverse the inhibition of lymphocyte proliferation induced by histamine and to increase the number of TIL in colorectal and gastric cancer patients [36, 40, 76]. Increased TIL was associated with prognostic significance in these trials, and is also considered significant in a range of other cancer types, including breast, ovarian, brain, and head and neck cancers.
Cell adhesion
CIM has been shown to have an inhibitory effect on cancer cell adhesion to endothelial cells independent of its H2RA activity. Using a monolayer cell adhesion assay the adhesion of HT-29 colorectal cancer cells to human umbilical vein endothelial cells was investigated for CIM and two other H2RAs (famotidine and ranitidine). Where CIM inhibited adhesion in a dose-dependent manner, the other H2RAs had no effect. In a nude mouse model, CIM dose-dependently reduced the incidence of HT-29 liver metastases, suppressing it completely at the highest dose (200 mg/kg/day) [83]. The effect on cell adhesion was mediated by the interaction between tumour sialyl Lewis antigens and E-selectin expressed on the endothelium.
Subsequent investigation has shown that there is a positive correlation between response to CIM treatment in colorectal cancer patients and high expression levels of sialyl Lewis-X and sialyl Lewis-A [39]. The reported 10-year cumulative survival rate of the CIM group with higher staining of sialyl Lewis-X in tumours was 95.5%, whereas that of a control group was 35.1% (P = 0.0001).
In addition to colorectal cancer, the inhibitory effect on cell adhesion has been demonstrated for other cancers, including breast [84], salivary gland tumours [85], gastric cancer [86] and glioblastoma [31].
Angiogenesis
The final mechanism of action that has been investigated in relation to the anti-cancer action of CIM is the effect it has on tumour neo-angiogenesis. Ghosh et al investigated the role of histamine in the production of vascular endothelial growth factor (VEGF) in carrageenin-induced granulation tissue in rats, and found that it was mediated by the H2 receptor, and that the upregulation of VEGF induced by histamine was reversed by CIM [87]. A study comparing CIM and roxatidine (another H2RA), found that both drugs strongly reduced colon 38 tumour implants in C57BL /6 mice syngeneic mice, and that this inhibition was related to reduced expression of VEGF and reduced micro-vessel density in the implanted tumours [88]. Additionally, there is also evidence that the anti-angiogenic effect of CIM administration may also be related to a reduced expression of platelet-derived endothelial growth factor (PDECGF), as well as VEGF, in mouse and rat models of bladder cancer [89].
Mechanistically, it has been suggested that VEGF expression is increased by histamine via the activation of the cyclooxygenase-2 (COX-2) pathway in colorectal cancer cell lines, a process mediated by the H2 and H4 receptors [90]. This process was disrupted by the H2RA zolantidine and H4RA JNJ 7777120. In contrast, CIM showed no effect on VEGF expression in an in vitro endothelial cell model of angiogenesis [91].
Our take
Next steps
The abundance of clinical evidence shows that CIM has demonstrable therapeutic effects in a range of cancers, particularly cancers of the gastrointestinal tract, RCC, and melanoma (summarised in Table 1). There is also evidence, both in vitro and in vivo, that these effects are most likely related to well-documented immunomodulatory effects. Furthermore, the evidence indicates that these effects may extend beyond the direct effect on the H2 histamine receptor and that CIM has off-target effects which are not shared by other H2RAs. We can hypothesise, therefore, that a portion of the variability of response to CIM reported in different clinical trials may be explained by the degree of variability of immune function in cancer patients. In common with other immunotherapeutic agents, this suggests that CIM may be more efficacious in patients with lower tumour burden and higher immune function, and in cancers with a greater antigenic potential. Indeed, an explanation proffered for the differences in response reported by different clinical trials in RCC was the better performance status (related to tumour burden) in patients in early trials compared to the poorer response reported in later trials [58].
This suggests that, in general, CIM should not be used as a single agent in an adjuvant setting, or in patients with large tumour burden or in cancer types which are known not to respond well to immunotherapeutic intervention. However, with that proviso, there is still considerable scope for clinical investigation of CIM as an immunostimulant, with a possible anti-metastatic action, in a range of cancer types.
In particular, one window of opportunity exists in using CIM to address the issue of post-operative immunosuppression. It is known that surgical resection, a mainstay of cancer treatment for many forms of the disease, causes a post-surgical immune suppression that may be associated with an increased risk of recurrence or metastatic spread [92, 93]. There is already strong evidence, summarised in a Cochrane Review, that perioperative CIM is associated with reduced immunosuppression and a lower risk of disease recurrence in the curative resection of colorectal cancer [41]. Moreover, the increased risk of post-surgical recurrence exists in other forms of cancer, including breast, lung, head and neck, and osteosarcoma. In breast cancer, for example, the perioperative use of the non-steroidal anti-inflammatory drug (NSAID) ketorolac is being investigated as potential agent to improve survival following mastectomy [94]. CIM is of potential benefit in these other cancers, in addition to the established benefit in colorectal cancer.
Table 1. Summary of evidence by cancer type.
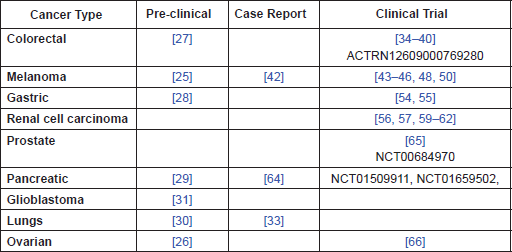
Given the evidence that perioperative CIM reduces post-surgical immunosuppression, it is suggested that there is a need for clinical trials to establish whether it may be of benefit, in terms of overall survival, in the following cancer types:
• Colorectal
• Breast
• Non-small Cell Lung Cancer
• Osteosarcoma
• Ovarian
• Pancreatic
It may be critical in these trials to start CIM treatment in the days immediately prior to surgery and continue for a number of weeks following, indeed it is worth noting that the most impressive clinical trial data show a dramatically improved survival for colorectal patients treated with oral CIM (800 mg/day) and oral 5-FU (200 mg/day)—the CIM-treated group had 10-year survival of 84.6% versus 49.8% for the 5-FU-only group [39]. Additionally, the investigation into the combined perioperative use of CIM and diclofenac/ketorolac warrants attention.
There is evidence that an immunological adjuvant may of benefit in a wide range of cancers, including some of those in which CIM has already shown some clinical benefit:
• Melanoma
• RCC
• Gastric cancer
• Glioblastoma
It is suggested that CIM be investigated as an adjuvant to the existing standard of care therapies in these diseases.
Given the primary putative mechanisms of action—the strongest evidence is for effects on immunity and cell adhesion—there are a number of additional agents that warrant investigation for synergy with CIM, some of which are listed in the supplementary material accompanying this paper.
New protocols
It is instructive to review the ongoing clinical trials of CIM as an anti-cancer agent as they serve as useful templates for future investigations. The Australian trial of perioperative CIM in colorectal cancer (ACTRN12609000769280) builds directly on a number of similar earlier trials, which are effectively summarised in a Cochrane Review [41]. It is to be hoped that a positive result in this trial will focus attention once more on the potential of CIM to positively affect overall survival. Of note a recent analysis of the long-term results of the EORTC 22921 trial found that adjuvant fluorouracil-based chemotherapy after preoperative radiotherapy (with or without chemotherapy) does not affect disease-free or overall survival [95], suggesting that this is an indication where progress is urgently needed and where CIM already has shown strong clinical evidence of effect.
However, using CIM to address post-surgical immune suppression is not the only possible model of use. The other clinical trials on-going combine CIM with a number of other low-cost agents to form the novel drug combination TL-118. In this model of use, it is the combination of multiple repurposed drugs, with similar low toxicity and low costs, which together form effective and novel treatment options. In this manner, we can create multi-targeted protocols which pose minimal risks to patients and yet offer hope of therapeutic efficacy. A number of these protocols are described in the supplementary material. Of necessity, such combinations are speculative, and though the evidence for the individual agents may be strong, the evidence for these combinations is often mechanistic or based on pre-clinical data only. While there is a need for more pre-clinical studies, it can be argued that given the urgency of patient need and the low toxicity of these proposed combinations, it is acceptable that small patient trials will begin in the near future.
Conclusion
The evidence for an anti-cancer effect of CIM treatment comes from in vitro, in vivo, and considerable amounts of human data. There are a number of well-described mechanisms of action, particularly of multiple immunomodulatory effects which have been assessed in data from clinical trials as well as from in vivo models. As an agent CIM has well-established pharmacokinetics and an excellent toxicity profile. Its use in clinical trials together with several chemotherapeutic agents has not shown any clinically relevant interactions, except with epirubicin, and showed evidence of a possible protective effect with vinorelbine and cisplatin. It is, therefore, a very strong candidate for repurposing as an oncological treatment, particularly as a perioperative treatment for surgical resection of solid tumours, in combination with existing standard treatments and alongside other repurposed drugs in a range of cancers.
A number of these multi-drug combinations have been outlined in the supplementary material in the hope that clinicians act upon this data to initiate clinical trials as a matter of some urgency.
Author contributions
Primary author: Pan Pantziarka. Contributing authors: Gauthier Bouche, Lydie Meheus, Vidula Sukhatme and Vikas P Sukhatme. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests. All the authors are associated with not-for-profit organisations that aim to repurpose drugs for oncology treatments.
References
1. Sabesin SM (1993) Safety issues relating to long-term treatment with histamine H2-receptor antagonists Aliment Pharmacol Ther 7 Suppl 2 35–40 PMID: 8103374
2. Chandrasekhara KL, Iyer SK and Macchia RJ (1981) Leucopenia and thrombocytopenia with cimetidine J Natl Med Assoc 73(2) 92, 98 PMID: 7205981 PMCID: 2552636
3. Joint Formulary Committee (2013) British National Formulary 66th edn (BMJ Group and Pharmaceutical Press)
4. Bodemar G, Norlander B and Walan A (1981) Pharmacokinetics of cimetidine after single doses and during continuous treatment Clin Pharmacokinet 6(4) 306–15 DOI: 10.2165/00003088-198106040-00005 PMID: 7249489
5. Bodemar G et al (1979) The absorption of cimetidine before and during maintenance treatment with cimetidine and the influence of a meal on the absorption of cimetidine–studies in patients with peptic ulcer disease Br J Clin Pharmacol 7(1) 23–31 DOI: 10.1111/j.1365-2125.1979.tb00892.x PMID: 760739 PMCID: 1429608
6. Martínez C et al (1999) Comparative in vitro and in vivo inhibition of cytochrome P450 CYP1A2, CYP2D6, and CYP3A by H2-receptor antagonists Clin Pharmacol Ther 65(4) 369–76 DOI: 10.1016/S0009-9236(99)70129-3 PMID: 10223772
7. Michalets EL (1998) Update: clinically significant cytochrome P-450 drug interactions Pharmacotherapy 18(1) 84–112 PMID: 9469685
8. Murray LS et al (1998) The effect of cimetidine on the pharmacokinetics of epirubicin in patients with advanced breast cancer: preliminary evidence of a potentially common drug interaction Clin Oncol (R Coll Radiol) 10(1) 35–8 DOI: 10.1016/S0936-6555(98)80109-X
9. Bekhti A and Pirotte J (1987) Cimetidine increases serum mebendazole concentrations. Implications for treatment of hepatic hydatid cysts Br J Clin Pharmacol 24(3) 390–2 DOI: 10.1111/j.1365-2125.1987.tb03186.x PMID: 3663452 PMCID: 1386263
10. Teshima T et al (1990) Radiation therapy of the para-aortic lymph nodes in carcinoma of the uterine cervix: the concurrent use of cimetidine to reduce acute and subacute side effects from radiation Clin Ther 12(1) 71–7 PMID: 2183942
11. Vassilomanolakis M et al (2001) Prevention of vinorelbine phlebitis with cimetidine. A two-step design study Suppor Care Cancer 9(2) 108–11 DOI: 10.1007/s005200000190
12. Sprowl JA et al (2013) Conjunctive therapy of cisplatin with the OCT2 inhibitor cimetidine: influence on antitumor efficacy and systemic clearance Clini Pharmacol Ther 94(5) 585–92 DOI: 10.1038/clpt.2013.145
13. Buttle GA et al (1962) Histamine formation and tumor growth Br Journal Cancer 16 131–40 DOI: 10.1038/bjc.1962.13
14. Burtin C et al (1981) Increased tissue histamine in tumour-bearing mice and rats Br J Cancer 43(5) 684–8 DOI: 10.1038/bjc.1981.99 PMID: 7248152 PMCID: 2010678
15. Burtin C et al (1983) Decreased blood histamine levels in patients with solid malignant tumours Br J Cancer 47(3) 367–72 DOI: 10.1038/bjc.1983.55 PMID: 6830687 PMCID: 2011313
16. Tutton PJ and Barkla DH (1978) Stimulation of cell proliferation by histamine H2 receptors in dimethylhdrazine-induced adenocarcinomata Cell Biol Int Rep 2(2) 199–202 DOI: 10.1016/0309-1651(78)90043-7 PMID: 667962
17. Burtin C et al (1982) Decrease in tumour growth by injections of histamine or serotonin in fibrosarcoma-bearing mice: influence of H1 and H2 histamine receptors Br J Cancer 45(1) 54–60 DOI: 10.1038/bjc.1982.7 PMID: 7059465 PMCID: 2010957
18. Gifford RR, Ferguson RM and Voss BV (1981) Cimetidine reduction of tumour formation in mice Lancet 1(8221) 638–40 DOI: 10.1016/S0140-6736(81)91555-5 PMID: 6110865
19. Osband ME et al (1981) Successful tumour immunotherapy with cimetidine in mice Lancet 1(8221) 636–8 DOI: 10.1016/S0140-6736(81)91554-3 PMID: 6110864
20. Gifford RR, Voss BV and Ferguson RM (1981) Cimetidine protection against lethal tumor challenge in mice Surgery 90(2) 344–51 PMID: 6454981
21. Dorr RT and Alberts DS (1982) Cimetidine enhancement of cyclophosphamide antitumour activity Br J cancer 45(1) 35–43 DOI: 10.1038/bjc.1982.5 PMID: 7059463 PMCID: 2010952
22. Hannant D et al (1982) Cimetidine and therapy of rodent tumours Br J Cancer 45(4) 613–4 DOI: 10.1038/bjc.1982.98 PMID: 7073951 PMCID: 2010991
23. Collins M and Hellmann K (1982) Histamine receptor antagonism and anti-tumour activity Br J Cancer 46(5) 817–20 DOI: 10.1038/bjc.1982.276 PMID: 6128996 PMCID: 2011159
24. Nordlund JJ and Askenase PW (1983) The effect of histamine, antihistamines, and a mast cell stabilizer on the growth of cloudman melanoma cells in DBA/2 mice J Invest Dermatol 81(1) 28–31 DOI: 10.1111/1523-1747.ep12538356 PMID: 6863977
25. Szincsák N et al (2002) Different h2 receptor antihistamines dissimilarly retard the growth of xenografted human melanoma cells in immunodeficient mice Cell Biol Int 26(9) 833–6 DOI: 10.1016/S1065-6995(02)90934-0 PMID: 12377215
26. Kikuchi Y et al (1985) Effects of cimetidine on tumor growth and immune function in nude mice bearing human ovarian carcinoma J Natl Cancer Inst 74(2) 495–8 PMID: 3871872
27. Adams WJ, Lawson JA and Morris DL (1994) Cimetidine inhibits in vivo growth of human colon cancer and reverses histamine stimulated in vitro and in vivo growth Gut 35(11) 1632–6 DOI: 10.1136/gut.35.11.1632 PMID: 7828988 PMCID: 1375627
28. Jiang C-G et al (2010) Cimetidine induces apoptosis in gastric cancer cells in vitro and inhibits tumor growth in vivo Oncol Rep 23(3) 693–700 PMID: 20127008
29. Sürücü O et al (2004) Tumour growth inhibition of human pancreatic cancer xenografts in SCID mice by cimetidine Inflamm Res 53 Suppl 1 S39–40 PMID: 15054609
30. Zheng Y et al (2013) Cimetidine suppresses lung tumor growth in mice through proapoptosis of myeloid-derived suppressor cells Mol Immunol 54(1) 74–83 DOI: 10.1016/j.molimm.2012.10.035
31. Lefranc F et al (2005) Combined cimetidine and temozolomide, compared with temozolomide alone: significant increases in survival in nude mice bearing U373 human glioblastoma multiforme orthotopic xenografts J Neurosurg 102(4) 706–14 DOI: 10.3171/jns.2005.102.4.0706 PMID: 15871514
32. Pantziarka P et al (2014) Repurposing Drugs in Oncology (ReDO)-mebendazole as an anti-cancer agent Ecancermedicalscience 8 443 PMID: 25075217 PMCID: 4096024
33. Armitage JO and Sidner RD (1979) Antitumour effect of cimetidine Lancet 1(8121) 882–3 DOI: 10.1016/S0140-6736(79)91306-0 PMID: 86136
34. Adams WJ et al (1994) Cimetidine preserves non-specific immune function after colonic resection for cancer Aust N Z J Surg 64(12) 847–52 DOI: 10.1111/j.1445-2197.1994.tb04562.x PMID: 7980260
35. Adams WJ and Morris DL (1994) Short-course cimetidine and survival with colorectal cancer Lancet 344(8939-8940) 1768–9 DOI: 10.1016/S0140-6736(94)92907-6 PMID: 7997018
36. Adams WJ and Morris DL (1997) Pilot study–cimetidine enhances lymphocyte infiltration of human colorectal carcinoma: results of a small randomized control trial Cancer 80(1) 15–21 DOI: 10.1002/(SICI)1097-0142(19970701)80:1<15::AID-CNCR3>3.0.CO;2-E PMID: 9210704
37. Svendsen LB et al (1995) Cimetidine as an adjuvant treatment in colorectal cancer. A double-blind, randomized pilot study Dis Colon Rectum 38(5) 514–8 DOI: 10.1007/BF02148852 PMID: 7736883
38. Kelly MD et al (1999) Randomized trial of preoperative cimetidine in patients with colorectal carcinoma with quantitative assessment of tumor-associated lymphocytes Cancer 85(8) 1658–63 DOI: 10.1002/(SICI)1097-0142(19990415)85:8<1658::AIDCNCR3>3.0.CO;2-Q PMID: 10223557
39. Matsumoto S et al (2002) Cimetidine increases survival of colorectal cancer patients with high levels of sialyl Lewis-X and sialyl Lewis-A epitope expression on tumour cells Br J Cancer 86(2) 161–7 DOI: 10.1038/sj.bjc.6600048 PMID: 11870500 PMCID: 2375187
40. Lin CY et al (2004) Perioperative cimetidine administration promotes peripheral blood lymphocytes and tumor infiltrating lymphocytes in patients with gastrointestinal cancer: Results of a randomized controlled clinical trial World J Gastroenterol 10 136–42 PMID: 14695785
41. Deva S and Jameson M (2012) Histamine type 2 receptor antagonists as adjuvant treatment for resected colorectal cancer Cochrane Database Syst Rev 8(8), p. CD007814 PMID: 22895966
42. Thornes RD, Lynch G and Sheehan MV (1983) Coumarin and cimetidine in malignant melanoma. Irish Medical Journal 76(1) 53 PMID: 6826342
43. Borgström S et al (1982) Human leukocyte interferon and cimetidine for metastatic melanoma N Engl J Med 307(17) 1080–1 DOI: 10.1056/NEJM198210213071716 PMID: 7121522
44. Flodgren P et al (1983) Metastatic malignant melanoma: regression induced by combined treatment with interferon [HuIFN-alpha(Le)] and cimetidine Int J Cancer 32(6) 657–65 DOI: 10.1002/ijc.2910320603 PMID: 6654521
45. Morton RF et al (1987) Phase II studies of single-agent cimetidine and the combination N-phosphonacetyl-L-aspartate (NSC-224131) plus L-alanosine (NSC-153353) in advanced malignant melanoma J Clin Oncol 5(7) 1078–82 PMID: 3598611
46. Creagan ET et al (1987) Three consecutive phase II studies of recombinant interferon alfa-2a in advanced malignant melanoma. Updated analyses Cancer 59(3 Suppl) 638–46 DOI: 10.1002/1097-0142(19870201)59:3 <638::AID-CNCR2820591312>3.0.CO;2-0 PMID: 10822463
47. Creagan ET et al (1990) Disseminated malignant melanoma and recombinant interferon: analysis of seven consecutive phase II investigations J Invest Dermatol 95(6 Suppl) 188S–192S DOI: 10.1111/1523-1747.ep12875512 PMID: 2124246
48. Schmidt H et al (2000) Subcutaneous interleukin-2 and interferon-alpha plus cisplatin with and without prophylactic cimetidine in patients with metastatic malignant melanoma: a phase II study Melanoma Res 10(1) 66–77 DOI: 10.1097/00008390200010010-00009 PMID: 10711642
49. Foster GR and Finter NB (1998) Are all type I human interferons equivalent? J Viral Hepat 5(3) 143–52 DOI: 10.1046/j.1365-2893.1998.00103.x PMID: 9658366
50. Mandanas R et al (1991) Phase II trial of cimetidine in metastatic melanoma. A Hoosier Oncology Group trial Am J Clin Oncol 14(5) 397–9 DOI: 10.1097/00000421-199110000-00007 PMID: 1951177
51. Arnot RS (1977) Cimetidine and gastric carcinoma Br Med J 2(6093) p. 1022 DOI: 10.1136/bmj.2.6093.1022-a PMID: 922362 PMCID: 1631768
52. Elder JB, Ganguli PC and Gillespie IE (1979) Cimetidine and gastric cancer Lancet 1(8124) 1005–6 DOI: 10.1016/S0140-6736(79)92757-0 PMID: 86721
53. Colin-Jones DG et al (1991) Post-cimetidine surveillance for up to ten years: incidence of carcinoma of the stomach and oesophagus Q J Med 78(285) 13–9 PMID: 1670060
54. Tønnesen H et al (1988) Effect of cimetidine on survival after gastric cancer Lancet 2(8618) 990–2 DOI: 10.1016/S0140-6736(88)90743-X PMID: 2902494
55. Langman MJS et al (1999) Prospective, double-blind, placebo-controlled randomized trial of cimetidine in gastric cancer. British Stomach Cancer Group Br J Cancer 81 1356–1362 DOI: 10.1038/sj.bjc.6690457 PMID: 10604733 PMCID: 2362962
56. Marshall ME et al (1987) Treatment of metastatic renal cell carcinoma with coumarin (1, 2-benzopyrone) and cimetidine: a pilot study J Clin Oncol 5(6) 862–6 PMID: 3585442
57. Herrmann R et al (1990) Phase II trial of coumarin and cimetidine in advanced renal cell carcinoma Ann Oncol 1(6) 445–6 PMID: 2083188
58. Venook AP, Davenport Y and Tseng A (1989) Activity of coumarin and cimetidine in metastatic renal cell carcinoma J Clin Oncol 7(3) 402–3 PMID: 2918335
59. Kinouchi T et al (1997) Treatment of advanced renal cell carcinoma with a combination of human lymphoblastoid interferon-alpha and cimetidine J Urol 157(5) 1604–7 DOI: 10.1016/S0022-5347(01)64806-7 PMID: 9112486
60. Kinouchi T et al (2006) Prospective randomized trial of natural interferon-alpha versus natural interferon-alpha plus cimetidine in advanced renal cell carcinoma with pulmonary metastasis J Cancer Res Clin Oncol 132(8) 499–504 DOI: 10.1007/s00432-006-0095-7 PMID: 16586071
61. Tatokoro M et al (2011) Phase-II trial of combination treatment of interferon-α, cimetidine, cyclooxygenase-2 inhibitor and renin-angiotensin-system inhibitor (I-CCA therapy) for advanced renal cell carcinoma Cancer Sci 102(1) 137–143 DOI: 10.1111/j.1349-7006.2010.01756.x
62. Inhorn L et al (1992) High-dose cimetidine for the treatment of metastatic renal cell carcinoma. A Hoosier Oncology Group study Am J Clin Oncol 15(2) 157–9 DOI: 10.1097/00000421-199204000-00012 PMID: 1553905
63. Bowrey PF et al (2000) Histamine, mast cells and tumour cell proliferation in breast cancer: does preoperative cimetidine administration have an effect? Br J Cancer 82(1) 167–70 DOI: 10.1054/bjoc.1999.0895 PMID: 10638985 PMCID: 2363200
64. Breuer S et al (2013) TL-118-anti-angiogenic treatment in pancreatic cancer: a case report Med Oncol 30(2) 585 DOI: 10.1007/s12032-013-0585-9 PMID: 23609193
65. Marshall ME, Butler K and Hermansen D (1990) Treatment of hormone-refractory stage D carcinoma of prostate with coumarin (1,2-benzopyrone) and cimetidine: a pilot study Prostate 17(2) 95–9 DOI: 10.1002/pros.2990170203 PMID: 2399194
66. Niwa K et al (2008) Prognostic implications of cimetidine on advanced serous ovarian carcinoma related to cyclooxygenase-2 expression Mol Med Rep 1(1) 119–22 PMID: 21479387
67. Smith TJ and Kaplowitz LG (1991) Pilot study of cimetidine in the treatment of Kaposi’s sarcoma in patients with acquired immunodeficiency syndrome J Nat Cancer Inst 83(2) 139–41 DOI: 10.1093/jnci/83.2.139-a PMID: 1988689
68. Jameson MB et al (2014) A randomized, placebo-controlled, double-blind phase II trial of peri-operative cimetidine in early colorectal cancer J ClinOncol 32(suppl 3) p. abstr 509
69. Medina VA and Rivera ES (2010) Histamine receptors and cancer pharmacology Br J Pharmacol 161(4) 755–67 DOI: 10.1111/j.1476-5381.2010.00961.x PMID: 20636392 PMCID: 2992892
70. Kennedy L et al (2012) Histamine and histamine receptor regulation of gastrointestinal cancers Transl Gastrointest Cancer 1(3) 215–227 PMID: 24639917 PMCID: 3955103
71. Watson SA et al (1993) Effect of histamine on the growth of human gastrointestinal tumours: reversal by cimetidine Gut 34(8) 1091–6 DOI: 10.1136/gut.34.8.1091 PMID: 8174960 PMCID: 1374360
72. Hahm KB et al (1996) Comparison of antiproliferative effects of 1-histamine-2 receptor antagonists, cimetidine, ranitidine, and famotidine, in gastric cancer cells Int J Immunopharmacol 18(6–7) 393–9 DOI: 10.1016/S0192-0561(96)00044-6 PMID: 9024941
73. Rajendra S et al (2004) The effect of H2 antagonists on proliferation and apoptosis in human colorectal cancer cell lines Dig Dis Sci 49(10) 1634–40 DOI: 10.1023/B:DDAS.0000043377.30075.ac PMID: 15573918
74. Mackenzie GG, Huang L, Alston N and Ouyang N et al (2013) Targeting mitochondrial STAT3 with the novel phospho-valproic acid (MDC-1112) inhibits pancreatic cancer growth in mice. Schneider, G. (ed.) PloS one 8(5) e61532 DOI: 10.1371/journal.pone.0061532 PMID: 23650499 PMCID: 3641121
75. O’Mahony L, Akdis M and Akdis CA (2011) Regulation of the immune response and inflammation by histamine and histamine receptors J Allergy Clin Immunol 128(6) 1153–62 DOI: 10.1016/j.jaci.2011.06.051
76. Li Y et al (2005) Effects of perioperative cimetidine administration on peripheral blood lymphocytes and tumor infiltrating lymphocytes in patients with gastrointestinal cancer: results of a randomized controlled clinical trial Hepatogastroenterology 52(62) 504–8 PMID: 15816467
77. Martin RK et al (2014) Mast cell histamine promotes the immunoregulatory activity of myeloid-derived suppressor cells J Leukoc Biol 96(July) 1–9 DOI: 10.1189/jlb.5A1213-644R PMID: 24610880
78. Elenkov IJ et al (1998) Histamine potently suppresses human IL-12 and stimulates IL-10 production via H2 receptors J Immunol 161(5) 2586–93 PMID: 9725260
79. Hegyesi H et al (2007) Impact of systemic histamine deficiency on the crosstalk between mammary adenocarcinoma and T cells J Pharmacol Sci 105(1) 66–73 DOI: 10.1254/jphs.FP0070636 PMID: 17895589
80. Lindau D et al (2013) The immunosuppressive tumour network: myeloid-derived suppressor cells, regulatory T cells and natural killer T cells Immunology 138(2) 105–15 DOI: 10.1111/imm.12036 PMCID: 3575763
81. Kubota T et al (2002) Cimetidine modulates the antigen presenting capacity of dendritic cells from colorectal cancer patients Br J Cancer 86(8) 1257–61 DOI: 10.1038/sj.bjc.6600233 PMID: 11953882 PMCID: 2375332
82. Katoh J et al (1998) Cimetidine reduces impairment of cellular immunity after cardiac operations with cardiopulmonary bypass J Thorac Cardiovasc Surg 116(2) 312–8 DOI: 10.1016/S0022-5223(98)70132-1 PMID: 9699585
83. Kobayashi K et al (2000) Cimetidine inhibits cancer cell adhesion to endothelial cells and prevents metastasis by blocking E-selectin expression Cancer Res 60(14) 3978–84 PMID: 10919677
84. Bobek V et al (2003) Inhibition of adhesion breast cancer cells by anticoagulant drugs and cimetidine Neoplasma 50(2) 148–51 PMID: 12740651
85. Fukuda M, Kusama K and Sakashita H (2008) Cimetidine inhibits salivary gland tumor cell adhesion to neural cells and induces apoptosis by blocking NCAM expression BMC Cancer 8 376 DOI: 10.1186/1471-2407-8-376 PMID: 19091137 PMCID: 2635382
86. Liu F-R et al (2011) Cimetidine inhibits the adhesion of gastric cancer cells expressing high levels of sialyl Lewis x in human vascular endothelial cells by blocking E-selectin expression Int J Mol Med 27(4) 537–44 PMID: 21327325
87. Ghosh AK, Hirasawa N and Ohuchi K (2001) Enhancement by histamine of vascular endothelial growth factor production in granulation tissue via H(2) receptors Br J Pharmacol 134(7) 1419–28 DOI: 10.1038/sj.bjp.0704372 PMID: 11724747 PMCID: 1573073
88. Tomita K, Izumi K and Okabe S (2003) Roxatidine- and cimetidine-induced angiogenesis inhibition suppresses growth of colon cancer implants in syngeneic mice J Pharmacol Sci 93(3) 321–30 DOI: 10.1254/jphs.93.321 PMID: 14646250
89. Chihara Y et al (2009) Anti-tumor effect of cimetidine via inhibiting angiogenesis factors in N-butyl-N-(4-hydroxybutyl) nitrosamine-induced mouse and rat bladder carcinogenesis Oncol Rep 22(1) 23–8 DOI: 10.3892/or_00000401 PMID: 19513500
90. Cianchi F et al (2005) The role of cyclooxygenase-2 in mediating the effects of histamine on cell proliferation and vascular endothelial growth factor production in colorectal cancer Clin Cancer Res 11(19 Pt 1) 6807–15 DOI: 10.1158/1078-0432.CCR-05-0675 PMID: 16203768
91. Lu Q et al (2013) Histamine synergistically promotes bFGF-induced angiogenesis by enhancing VEGF production via H1 receptor J Cell Biochem 114(5) 1009–19 DOI: 10.1002/jcb.24440
92. Shakhar G and Ben-Eliyahu S (2003) Potential prophylactic measures against postoperative immunosuppression: could they reduce recurrence rates in oncological patients? Ann Surg Oncol 10(8) 972–92 DOI: 10.1245/ASO.2003.02.007 PMID: 14527919
93. Forget P, Simonet O and De Kock M (2013) Cancer surgery induces inflammation, immunosuppression and neo-angiogenesis, but is it influenced by analgesics? F1000Res 2 102 PMID: 24358839 PMCID: 3752648
94. Forget P et al (2013) Perioperative ketorolac in high risk breast cancer patients. Rationale, feasibility and methodology of a prospective randomized placebo-controlled trial Med Hypotheses 81(4) 707–12 DOI: 10.1016/j.mehy.2013.07.033 PMID: 23937996
95. Bosset J-F et al (2014) Fluorouracil-based adjuvant chemotherapy after preoperative chemoradiotherapy in rectal cancer: long-term results of the EORTC 22921 randomised study The Lancet Oncol 15(2) 184–90 DOI: 10.1016/S1470-2045(13)70599-0
Appendix
Introduction
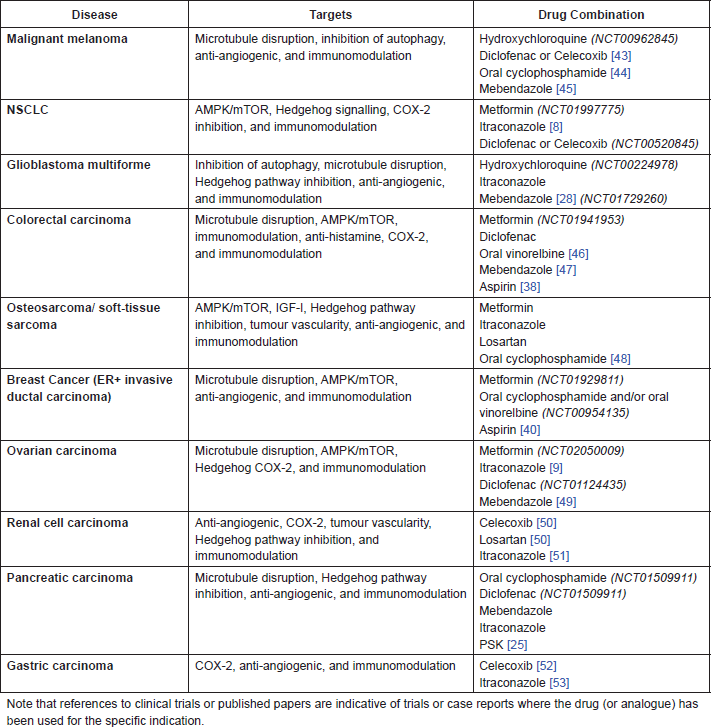
The following drugs warrant further investigation in combination with cimetidine (CIM), both in pre-clinical studies and potentially in clinical trials. These combinations, listed in Table A1, have been selected on the basis of existing pre-clinical and clinical experience in each of the indications. In some cases, these combinations replicate existing protocols currently being tested in clinical trials, but substitute known and repurposed drugs for the newer and/or more toxic agents currently being investigated. All these proposed combinations are expected to display relatively low toxicity and use low-cost and generally available agents.
Higher-priority agents
The agents listed below have a high degree of clinical evidence of efficacy and are currently either in clinical use in oncology or are currently being investigated in clinical trials. They have been selected as potential agents to be used in combination with CIM. Note that these drugs are not listed in order of priority.
• Metronomic chemotherapy—There is increasing clinical interest in using metronomic dosing schedules in a range of existing chemotherapeutic drugs, particularly cyclophosphamide, capecitabine, etoposide, temozolomide, and vinorelbine [1, 2]. At the continuous low doses used in metronomic chemotherapy, there is little evidence of a direct cytotoxic effect on tumour cells, with increasing evidence that the therapeutic effect is primarily driven by anti-angiogenic and immunomodulatory actions [3, 4]. Combining low-dose metronomic chemotherapy with other immunotherapeutic agents is an attractive proposition that is being actively investigated in clinical settings [5, 6]. Given the strong evidence that CIM acts in multiple immunomodulatory ways, there is every reason to believe that it would synergise with existing metronomic chemotherapy regimens to reverse cancer-induced immune suppression, improve activity of cytotoxic T lymphocytes, and prime immune responses in a Th1 direction. There is no evidence to suggest that the addition of CIM to these regimens would increase the lower levels of toxicity associated with metronomic chemotherapy.
• Itraconazole—This broad spectrum antifungal drug is being, or has been, clinically investigated as an anti-cancer agent in a number of trials, including for metastatic prostate cancer (NCT00887458), basal cell carcinoma [7], non-small cell lung cancer [8], refractory ovarian cancer [9], and triple-negative breast cancer [10]. The putative mechanisms of action are anti-angiogenic and inhibition of the Hedgehog signalling pathway [11, 12]. There is a pre-clinical evidence to suggest that the combination of Hedgehog pathway inhibition and the targeting of myeloid-derived suppressor cells (MSDC) may be a beneficial strategy in some hard to treat tumours such as pancreatic cancer [13]. It is suggested, therefore, that the combination of itraconazole and CIM be investigated in pancreatic and other solid tumours. It should be noted that there is some evidence in animal models of an interaction between itraconazole and CIM, such that the AUC of CIM was increased by 25%, an effect which may be of clinical benefit in extending the therapeutic effect of this drug combination [14].
• Diclofenac/Ketorolac—As with a number of other NSAID (particularly those with evidence of COX-2 inhibitory properties), diclofenac shows some evidence of anti-cancer activity. There is evidence that perioperative or intraoperative diclofenac or ketorolac may be associated with lower risk of cancer recurrence or metastatic spread following surgical resection of tumours [15]. Additionally, there is pre-clinical evidence of a direct anti-cancer role of diclofenac in a number of different malignancies. For example, there is in vivo evidence in ovarian cancer [16], in melanoma [17], and glioblastoma [18]. Diclofenac has also been used in the treatment of desmoid tumours in adults and paediatric patients, including in combination with vinblastine [19]. Mechanisms of action of diclofenac include anti-inflammatory effects, inhibition of COX-2 and effects on tumour cell metabolism [17]. Of particular interest is the immunological effect of diclofenac, which has been shown, ex vivo, to potently reverse post-irradiation immunosuppression [20]. It is hypothesised that effects of diclofenac or ketorolac, including anti-inflammatory and pro- immunity effects, would synergise with the immunotherapeutic effects of CIM and that this combination warrants clinical investigation.
Table A1. Proposed drug combinations with CIM for specific indications.
• PSK/PSP—PSK (Polysaccharide K/Krestin) is a protein-bound polysaccharide isolated from the cultured mycelium of the CM-101 strain of the mushroom Coriolus versicolor, which has a long history of medicinal use in traditional Chinese medicine. PSP (Polysaccharopeptide) is a related compound from the COV-1 strain of Coriolus versicolor that is also used medicinally, although there has been a less clinical investigation of PSP compared to PSK [21]. PSK has been used clinically in Japan since the late 1970s, principally with chemotherapy for the post-operative treatment of gastric, colorectal, and small-cell lung cancers. Numerous clinical trials and meta-analyses have indicated that there is a statistically significant effect on overall and disease-free survival for patients treated with curative resection followed by adjuvant PSK in these three cancers, and these are effectively summarised by Maehara et al [22]. There have been fewer clinical trials evaluating the use of PSP, though it is notable that a small trial in non-small cell lung cancer patients found a slower disease progression than in non-treated controls [23]. Also notable is a double-blinded clinical trial in canine hemangiosarcoma which found that high-dose PSP significantly delayed the progression of metastases and afforded the longest survival times reported to date in this condition [24]. The anti-cancer activity of both PSK and PSP is understood to be primarily due to diverse effects on the immune system, including reversal of immune suppression, upregulation of Th1 cytokine production and direct effects on tumour cells. These mechanisms of action parallel those of CIM, suggestive of a possible synergy that should be investigated in particularly intractable malignancies, such as pancreatic cancer [25] or soft-tissue sarcomas.
• Mebendazole—This widely used anthelmintic drug has shown pre-clinical and clinical evidence of activity against a range of cancers. It is being investigated in two clinical trials, with temozolomide, in glioblastoma, and there are case reports in colorectal and adrenocortical carcinoma [26]. The primary mechanism of action as an antiparasitic is the inhibition of tubulin polymerisation in the gut of helminths, and microtubule disruption has also been assessed in relation to its potential anti-cancer activity, for example in a melanoma and glioblastoma models [27, 28]. There is some evidence that CIM increases the peak plasma levels of mebendazole [29], an effect that may prove therapeutically useful in combination protocols. In particular, both drugs show evidence of anti-cancer activity in colorectal cancers, and it is suggested that clinical or pre-clinical investigation for colorectal disease is warranted.
• Hydroxychloroquine—The anti-malarial drugs chloroquine and hydroxychloroquine are known inhibitors of autophagy currently being investigated with a range of standard of care treatments against cancer [30]. The rationale is that cellular stresses generated by cancer treatments induce an autophagic response from tumour cells and that this autophagic state confers resistance to treatment. Inhibition of autophagy in such cases blocks this resistance and increases radio- and chemo-sensitivity [31]. However, there is intriguing evidence that the effectiveness of autophagy inhibition as an effective strategy can be compromised by defective immune responses [32, 33]. Possibly this is because autophagy inhibition leads to greater immunogenic cell death (ICD) in certain treatment modalities, such as radiotherapy, photodynamic therapy and other ROS-related cell death pathways (though not necessarily for certain chemotherapeutic agents) [32, 34, 35], but in an immunosuppressive environment ICD does not lead to greater anti-tumour immune response. The use of CIM to reverse immunosuppression in parallel to autophagy inhibition, (with chloroquine, hydroxychloroquine or other inhibitor, such as the antibiotic clarithromycin [36]), could therefore be a fruitful strategy to pursue in a clinical trial.
• Aspirin—There is abundant epidemiological evidence of a cancer-preventative effect of long-term aspirin usage, summarised for example by Thun et al [37], in recent years there has also been an increasing interest in the adjuvant effects of aspirin treatment [38–40]. In particular the use of aspirin post-diagnosis has been shown to be associated with improved overall survival in colorectal, breast, prostate, and oesophago-gastric cancers [40, 41]. There is also some evidence that aspirin use is associated with improved long-term survival in non-small cell lung cancer treated with curative resection [42]. It is proposed, therefore, that aspirin be combined with CIM for long-term post-operative protocols in a number of cancer indications, including colorectal and breast cancer.
References
1. André N, Carré M and Pasquier E (2014) Metronomics: towards personalized chemotherapy? Nat Rev Clin Oncol 11(7) 413–31 DOI: 10.1038/nrclinonc.2014.89 PMID: 24913374
2. Lien K et al (2013) Low-dose metronomic chemotherapy: a systematic literature analysis Eur J Cancer 49(16) 3387–95 DOI: 10.1016/j.ejca.2013.06.038 PMID: 23880474
3. Nars MS and Kaneno R (2013) Immunomodulatory effects of low dose chemotherapy and perspectives of its combination with immunotherapy Int J cancer 132(11) 2471–8 DOI: 10.1002/ijc.27801
4. Hahnfeldt P, Folkman J and Hlatky L (2003) Minimizing long-term tumor burden: the logic for metronomic chemotherapeutic dosing and its antiangiogenic basis J Theor Biol 220(4) 545–54 DOI: 10.1006/jtbi.2003.3162 PMID: 12623285
5. Lasalvia-Prisco E et al (2012) Addition of an induction regimen of antiangiogenesis and antitumor immunity to standard chemotherapy improves survival in advanced malignancies Med Oncol 29(5) 3626–33 DOI: 10.1007/s12032-012-0301-1 PMID: 22810591 PMCID: 3505507
6. Soriano JL et al (2011) Metronomic cyclophosphamide and methotrexate chemotherapy combined with 1E10 anti-idiotype vaccine in metastatic breast cancer Int J Breast Cancer 2011(Mc) Article ID 710292 DOI: 10.4061/2011/710292
7. Kim DJ et al (2014) Open-label, exploratory phase II trial of oral itraconazole for the treatment of basal cell carcinoma J Clin Oncol pp. 1–7 DOI: 10.1200/JCO.2013.49.9525
8. Rudin CM et al (2013) Phase 2 study of pemetrexed and itraconazole as second-line therapy for metastatic nonsquamous non-small-cell lung cancer J Thorac Oncol 8(5) 619–23 PMID: 23546045 PMCID: 3636564
9. Tsubamoto H et al (2014) Impact of combination chemotherapy with itraconazole on survival of patients with refractory ovarian cancer Anticancer Res 34(5) 2481–7 PMID: 24778064
10. Tsubamoto H, Sonoda T and Inoue K (2014) Impact of itraconazole on the survival of heavily pre-treated patients with triple-negative breast cancer Anticancer Res 34(7) 3839–44 PMID: 24982411
11. Chong CR et al (2007) Inhibition of angiogenesis by the antifungal drug itraconazole ACS Chem Biol 2(4) 263–70 DOI: 10.1021/cb600362d PMID: 17432820
12. Antonarakis ES et al (2013) Repurposing itraconazole as a treatment for advanced prostate cancer: a noncomparative randomized phase II trial in men with metastatic castration-resistant prostate cancer Oncologist 18(2) 163–73 DOI: 10.1634/theoncologist.2012-314 PMID: 23340005 PMCID: 3579600
13. Hamada S, Masamune A and Shimosegawa T (2013) Novel therapeutic strategies targeting tumor-stromal interactions in pancreatic cancer Front Physiol 4 331 DOI: 10.3389/fphys.2013.00331 PMID: 24273517 PMCID: 3822297
14. Karyekar CS et al (2004) Renal interaction between itraconazole and cimetidine J Clin Pharmacol 44(8) 919–27 DOI: 10.1177/0091270004266783 PMID: 15286096
15. Forget P et al (2014) Intraoperative use of ketorolac or diclofenac is associated with improved disease-free survival and overall survival in conservative breast cancer surgery Br J Anaesth 1–6 DOI: 10.1093/bja/aet464 PMID: 24464611
16. Zerbini LF et al (2011) Combinatorial effect of non-steroidal anti-inflammatory drugs and NF-κB inhibitors in ovarian cancer therapy PloS One 6(9) e24285 DOI: 10.1371/journal.pone.0024285 PMCID: 3171406
17. Gottfried E et al (2013) New aspects of an old drug–diclofenac targets MYC and glucose metabolism in tumor cells PloS One 8(7) e66987 DOI: 10.1371/journal.pone.0066987 PMID: 23874405 PMCID: 3706586
18. Chirasani SR et al (2013) Diclofenac inhibits lactate formation and efficiently counteracts local immune suppression in a murine glioma model Int J Cancer 132(4) 843–53 DOI: 10.1002/ijc.27712
19. Lackner H et al (2004) Multimodal treatment of children with unresectable or recurrent desmoid tumors: an 11-year longitudinal observational study J Pediatr Hematol Oncol 26(8) 518–22 DOI: 10.1097/01.mph.0000130219.26284.b3 PMID: 15284591
20. Wasserman J et al (1989) Immunosuppression in irradiated breast cancer patients: in vitro effect of cyclooxygenase inhibitors Bull N Y Acad Med 65(1) 36–44 PMID: 2513994 PMCID: 1807786
21. Ng TB (1998) A review of research on the protein-bound polysaccharide (polysaccharopeptide, PSP) from the mushroom Coriolus versicolor (Basidiomycetes: Polyporaceae) Gen Pharmacol 30(1) 1–4 DOI: 10.1016/S0306-3623(97)00076-1 PMID: 9457474
22. Maehara Y et al (2012) Biological mechanism and clinical effect of protein-bound polysaccharide K (KRESTIN(®)): review of development and future perspectives Surg Today 42(1) 8–28 DOI: 10.1007/s00595-011-0075-7 PMCID: 3253283
23. Tsang KW et al (2003) Coriolus versicolor polysaccharide peptide slows progression of advanced non-small cell lung cancer Respir Med 97(6) 618–24 DOI: 10.1053/rmed.2003.1490 PMID: 12814145
24. Brown DC and Reetz J (2012) Single agent polysaccharopeptide delays metastases and improves survival in naturally occurring hemangiosarcoma Evid Based Complement Alternat Med eCAM DOI: 10.1155/2012/384301 PMCID: 3440946
25. Onishi H et al (2013) Protein-bound polysaccharide decreases invasiveness and proliferation in pancreatic cancer by inhibition of hedgehog signaling and HIF-1α pathways under hypoxia Cancer Lett 335(2) 289–98 DOI: 10.1016/j.canlet.2013.02.041 PMID: 23485726
26. Pantziarka P et al (2014) Repurposing drugs in oncology (ReDO)-mebendazole as an anti-cancer agent Ecancermedicalscience 8 443 PMID: 25075217 PMCID: 4096024
27. Doudican N, Rodriguez A, Osman I and Orlow SJ (2008) Mebendazole induces apoptosis via Bcl-2 inactivation in chemoresistant melanoma cells Mol Cancer Res 6(8) 1308–15 DOI: 10.1158/1541-7786.MCR-07-2159 PMID: 18667591
28. Bai R et al (2011) Antiparasitic mebendazole shows survival benefit in 2 preclinical models of glioblastoma multiforme Neuro Oncol 13(9) 974–82 DOI: 10.1093/neuonc/nor077 PMID: 21764822 PMCID: 3158014
29. Bekhti A and Pirotte J (1987) Cimetidine increases serum mebendazole concentrations. Implications for treatment of hepatic hydatid cysts Br J Clin Pharmacol 24(3) 390–2 DOI: 10.1111/j.1365-2125.1987.tb03186.x PMID: 3663452 PMCID: 1386263
30. Carew JS, Kelly KR and Nawrocki ST (2012) Autophagy as a target for cancer therapy: new developments. Cancer Manag Res 4 357–65 PMID: 23091399 PMCID: 3474143
31. Choi KS (2012) Autophagy and cancer Exp Mol Med 44(2) 109–20 DOI: 10.3858/emm.2012.44.2.033 PMID: 22257886 PMCID: 3296807
32. Ko A et al (2014) Autophagy inhibition radiosensitizes in vitro, yet reduces radioresponses in vivo due to deficient immunogenic signalling Cell Death Differ 21(1) 92–9 DOI: 10.1038/cdd.2013.124
33. Bugaut H et al (2013) Bleomycin exerts ambivalent antitumor immune effect by triggering both immunogenic cell death and proliferation of regulatory T cells PloS One 8(6) e65181 DOI: 10.1371/journal.pone.0065181 PMID: 23762310 PMCID: 3676388
34. Garg AD et al (2013) ROS-induced autophagy in cancer cells assists in evasion from determinants of immunogenic cell death Autophagy 9(9) 1292–307 DOI: 10.4161/auto.25399 PMID: 23800749
35. Michaud M et al (2011) Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice Science 334(6062) 1573–7 DOI: 10.1126/science.1208347 PMID: 22174255
36. Moriya S et al (2013) Macrolide antibiotics block autophagy flux and sensitize to bortezomib via endoplasmic reticulum stress-mediated CHOP induction in myeloma cells Int J Oncol 42(5) 1541–50 DOI: 10.3892/ijo.2013.1870 PMCID: 3661227
37. Thun MJ, Jacobs EJ and Patrono C (2012) The role of aspirin in cancer prevention Nat Rev Clin Oncol 9(5) 259–267 DOI: 10.1038/nrclinonc.2011.199 PMID: 22473097
38. Bastiaannet E et al (2012) Use of Aspirin postdiagnosis improves survival for colon cancer patients Br J Cancer 106(1532-1827 (Electronic)), 1564–70 DOI: 10.1038/bjc.2012.101 PMID: 22454078 PMCID: 3341868
39. Langley RE et al (2011) Aspirin and cancer: has aspirin been overlooked as an adjuvant therapy? Br J Cancer 105(8) 1107–13 DOI: 10.1038/bjc.2011.289 PMID: 21847126 PMCID: 3208483
40. Fraser DM et al (2014) Aspirin use and survival after the diagnosis of breast cancer: a population-based cohort study Br J Cancer 111(3) 623–7 DOI: 10.1038/bjc.2014.264 PMID: 24945997 PMCID: 4119969
41. Phillips I et al (2013) Aspirin as a treatment for cancer Clin Oncol 25(6) 333–5 DOI: 10.1016/j.clon.2013.03.001
42. Fontaine E et al (2010) Aspirin and non-small cell lung cancer resections: effect on long-term survival Eur J Cardiothorac Surg 38 21–26 DOI: 10.1016/j.ejcts.2010.01.015 PMID: 20359903
43. Bhatt RS et al (2010) A phase 2 pilot trial of low-dose, continuous infusion, or “metronomic” paclitaxel and oral celecoxib in patients with metastatic melanoma Cancer 116(7) 1751–6 DOI: 10.1002/cncr.24902 PMID: 20120033 PMCID: 2847062
44. Borne E et al (2010) Oral metronomic cyclophosphamide in elderly with metastatic melanoma Invest New Drugs 28(5) 684–9 DOI: 10.1007/s10637-009-9298-5
45. Doudican NA et al (2013) XIAP downregulation accompanies mebendazole growth inhibition in melanoma xenografts Anticancer Drugs 24(2) 181–8 DOI: 10.1097/CAD.0b013e32835a43f1
46. Briasoulis E et al (2009) Dose-ranging study of metronomic oral vinorelbine in patients with advanced refractory cancer Clin Cancer Res 15(20) 6454–61 DOI: 10.1158/1078-0432.CCR-09-0970 PMID: 19808873
47. Nygren P et al (2013) Repositioning of the anthelmintic drug mebendazole for the treatment for colon cancer J Cancer Res Clin Oncol 139(12) 2133–40 DOI: 10.1007/s00432-013-1539-5 PMID: 24135855 PMCID: 3825534
48. André N et al (2011) Pilot study of a pediatric metronomic 4-drug regimen Oncotarget 2(12) 960–5 PMID: 22156656 PMCID: 3282100
49. Mukhopadhyay T et al (2002) Mebendazole elicits a potent antitumor effect on human cancer cell lines both in vitro and in vivo Clin Cancer Res 8(9) 2963–9 PMID: 12231542
50. Tatokoro M et al (2011) Phase-II trial of combination treatment of interferon-α, cimetidine, cyclooxygenase-2 inhibitor and renin-angiotensin-system inhibitor (I-CCA therapy) for advanced renal cell carcinoma Cancer Sci 102(1) 137–143 DOI: 10.1111/j.1349-7006.2010.01756.x
51. Nacev BA et al (2011) The antifungal drug itraconazole inhibits vascular endothelial growth factor receptor 2 (VEGFR2) glycosylation, trafficking, and signaling in endothelial cells J Biol Chem 286(51) 44045–56 DOI: 10.1074/jbc.M111.278754 PMID: 22025615 PMCID: 3243534
52. Han X et al (2014) Effect of celecoxib plus standard chemotherapy on serum levels of vascular endothelial growth factor and cyclooxygenase-2 in patients with gastric cancer Biomed Rep 2(2) 183–187 PMID: 24649093 PMCID: 3917754
53. Song Z et al (2011) Sonic hedgehog pathway is essential for maintenance of cancer stem-like cells in human gastric cancer PloS One 6(3) e17687 DOI: 10.1371/journal.pone.0017687 PMID: 21394208 PMCID: 3048871