Acute myeloid leukaemia at an early age: Reviewing the interaction between pesticide exposure and KMT2A-rearrangement
Maria S Pombo-de-Oliveira, Francianne Gomes Andrade, Gisele Dallapicola Brisson, Filipe Vicente dos Santos Bueno, Ingrid Sardou Cezar and Elda Pereira Noronha
Pediatric Hematology-Oncology Program, Research Center, Instituto Nacional de Câncer (INCA), Rio de Janeiro 20231-050, Brazil
Correspondence to: Maria S Pombo-de-Oliveira. Email: mpombo@inca.gov.br
Abstract
Acute myeloid leukaemia (AML) in early childhood is characterised by a high frequency of recurrent genomic aberrations associated with distinct myeloid subtypes, clinical outcomes and pathogenesis. Genomic instability is the first step of pathogenic mechanism in early childhood AML. A sum of adverse events is necessary to the development of infant AML (i-AML), which includes latency of biochemical-molecular and cellular effects. Inherited genetic susceptibility associated with exposures to biotransformation substances can modulate the risk of DNA damage and it is a very important piece in the pathogenic puzzle. In this review, we have aimed to explore the chain of events in the time-points of the natural history of i-AML, which includes maternal exposures during pregnancy, the speculations about the formation of somatic mutations during foetal life and the secondary genomic aberrations associated with i-AML. The modulation of risk conferred by xenobiotic metabolism´s genes variants is the bottom line of the pathogenic process. Since we have conducted observational and molecular investigations in early childhood leukaemia, the data focused here is based on Brazilian findings with summarised results of our experience with epidemiological and molecular studies in early-age leukaemia.
Keywords: acute myeloid leukaemia, KMT2A-rearrangements, pesticides maternal exposure, gene polymorphisms
Copyright: © the authors; licensee ecancermedicalscience. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Published: 30/11/2017; Received: 03/05/2017
The profile of genomic aberrations found in early-age AML
Acute myeloid leukaemia (AML) accounts for about 20% of childhood leukaemia. There are increased incidence rates occurring in infants (less than 1–2 years of age, i-AML). The incidence declines dramatically in children between five and nine years of age at diagnosis, and then it increases again in adolescents and young adults [1].
AML is a heterogeneous disease that develops through the transformation of a haematopoietic progenitor cell as a result of a block in differentiation and uncontrolled proliferation, leading to an accumulation of immature cells and the decrease of mature blood cells. It was postulated that AML might be originated by mutations in genes involved in proliferative and signal transduction pathways, as well as transcription factors of progenitor myeloid cells [2, 3]. The major AML subtypes are originated from chromosomal rearrangements with fusion genes, as elegantly described by Balgobind et al [3]. In settings such as infant AML (i-AML), there are strong evidences that aberrant fusion genes initiate during foetal life, although the majority of the biological investigations were demonstrated in acute lymphoblastic leukaemia (ALL) samples [4].
The most frequent chromosomal abnormalities in children with AML (c-AML) older than two years are those associated with the core-binding factor (CBF) aberrations, such as t(8;21)(q22;q22)/RUNX1-RUNX1T1 and inv(16)(p13.1;q22)/CBFβ-MYH11. Acute promyelocytic leukaemia (APL) with t(15;17)(q22;q21)/PML-RARA is also frequent in children [5]. In recent analysis of i-AML series in Brazil, the RUNX1-RUNX1T1, CBFβ-MYH1 and KMT2A (or MLL) rearrangements (KMT2A-r) and PML-RARA occurred in frequencies of 12.2%, 17.4%, 67.6% and 3.2%, respectively [6]. Amongst infants, CBF-group and APL are rare, and the most common morphological subtypes are myelomonocytic (AML-M4/M5) and megakaryocytic leukaemia (AML-M7). The recurrent KMT2A-r is highly prevalent in our series of i-AML cases. Other chromosomal translocations, such as the t(7;12)(q36;p13)/MNX1-ETV6, t(8;16)(p11;p13)/MYST3-CREBBP, t(1;22)(p13;q13)/RBM15-MKL1 and inv(16)(p13.3q24.3)/CBFA2T3-GLIS2, are all specifically associated with i-AML in variable prevalent frequencies that encompass the remaining 30% of aberrations [7–11].
The great majority of i-AML clinically presents with high white blood cell counts, hepatosplenomegaly, often central nervous system involvement and chloroma. A subset of i-AML has pre-leukaemia or constitutional syndromes that suggest heterogeneous etiologies for these cases. Indeed, approximately 10% of i-AML has myelodysplastic syndrome with monosomy 7 or del(7q), has Down Syndrome (DS), has Noonan syndrome or carry neurofibromatosis type 1 mutations, which all predispose to early AML [12, 13]. Children with DS have an increased risk of developing leukaemia, and among neonates and infants, AML is associated with GATA-1 recurrent mutations [14–16]. GATA-1 gene encodes a transcription factor and the mutation results in a truncated form of the protein GATA-1 that in physiological haematopoiesis interacts with other myeloid-lineage regulator genes. GATA-1mutations associated with trisomy 21 are crucial events to initiate megakaryoblastic abnormal proliferation [14, 15]. Evidence shows that likewise KMT2A-r, the GATA-1 mutation occurs during foetal life [17]. GATA-1 mutation is present in transient abnormal myelopoiesis (TAM), a clonal pre-leukaemia condition that occurs in about 10% of neonates with DS, presenting at a median age of 3–7 days with the accumulation of megakaryoblasts, increased leukocytes and thrombocytopenia. The majority of TAM regresses spontaneously without treatment; however, about 20% of these patients will develop megakaryocytic leukaemia in the first four years of life [18, 19]. This group of patients is often excluded in clinical and epidemiological studies that explore risk factors associated with etiopathogenesis of AML.
Since we have performed clinical, epidemiological and molecular studies in early-age childhood leukaemia, this review focuses on the development and data from maternal exposures during pregnancy and molecular events associated with i-AML (children with less than 24 months, excluding AML associated with Down Syndrome).
Therapy-related myeloid leukaemia as proxy for maternal exposures during pregnancy associated with early molecular events in infant myeloid leukaemia
Given the young age of most children with i-AML, it was proposed and it is consensually accepted that early-age leukaemia is initiated before the birth of the child. The main evidence for a prenatal origin of childhood leukaemia was described in identical twins with concordant leukaemia. The same genomic fusion gene sequences were identified in twin pairs with acute leukaemia, which led to the assumption of a common clonal origin. The plausible basis for this finding is that in one twin foetus, clonal progeny spread to the co-twin via the vascular net. This assumption was endorsed by the identification of clonotypic gene fusion sequences, such as KTM2A-AFF1 and ETV6-RUNX1, in dried neonatal blood spots of children who subsequently developed leukaemia [20, 21]. It is clear that acquired genetic abnormalities, especially those gene fusions are the most frequently found genetic alterations in early infancy (< 24 months of age). It is unlikely, however, that a single chromosome translocation or gene mutation itself would be enough to cause overt leukaemia, and alterations in other classes of genes that impair cell differentiations (RAS-MAPkinase) probably cooperate with the survival of a malignant clone. Deregulation of the MAPK genes signalling caused by somatic mutations and particularly RAS mutation was associated with i-AML and chemical exposures [2, 3].
Based on biologic similarities between the therapy-related myeloid neoplasm (t-MN) and infant leukaemia with similar KMT2A-r caused by topoisomerase II (Topo-II) inhibitors [22], the t-MN leukemogenesis could be proxy to modelling studies associated with biochemical-molecular events in the early stage of i-AML with KMT2A-r. In t-MN investigations, the dose-effect associations can be well documented with qualitative and quantitative measurements, while in i-AML, the associations’ estimates are a presumption of biological plausibility.
Patients with a t-MN have clonal genomic abnormalities in their bone marrow cells, which frequently are correlated with the type of preceding cytotoxic compound, the amount of drug exposed and the latency period. The classical t-MNs occurred after exposure to alkylating agents, Topo-II inhibitors and/or radiation therapy, and were described after a latency period of 5–10 years, often preceded by a myelodysplastic phase [22]. Nowadays, several reports on t-MN have unraveled the multistep process, suggesting that drugs interfering with DNA remodelling by Topo-II inhibitors can mediate the formation of specific chromosomal translocation breakpoints. Chemotherapy agents targeting Topo-II enzymes increase the steady-state levels of cleavage complexes by preventing religation of the transient Topo-II break, eventually leading to apoptosis and cell death. It increases the chances of DNA lesions being repaired by nonhomologous end joining, leading to chromosomal translocations (mainly the 11q23 region). Chemotherapy used to treat primary malignancies, in particular, Topo-II inhibitors, leading to specific gene mutations, creates a genetic frame on which traditional acute leukaemia chemotherapy only selects resistant clones [23, 24]. The analysis of KTM2A genomic breakpoint junction sequences has shown duplicated regions up to several hundred bases long from KTM2A and/or its partner gene on either derivative chromosomes, or deletions of several hundred bases. Topo-II creates 4-base staggered double-stranded breaks (DSB) in DNA, but also introduces single-stranded nicks as kinetic intermediates of DSB. The precision of the breakpoint junction sequences and the results of DNA Topo-II cleavage assays in treatment-related leukaemia suggest a mechanism in which two DNA Topo-II introduce separate single-stranded nicks in duplex DNA that are staggered by up to several hundred bases. Subsequent template-directed polymerisation of the single-stranded overhangs between the staggered nicks would then generate the sequence duplications. This leads to a DNA damage-repair model in which various naturally occurring DNA Topo-II poisons induce DNA Topo II-mediated damage in leukaemia pathogenesis [25, 26].
One interesting observation concerns the latency of t-MN and early-age leukaemia. The latency period to develop t-MN is much shorter for KTM2A-r than the latency period of t-MN with unbalanced aberrations, for example, monosomy-5 or monosomy-7 [27]. Important to mention that the clustered oxidative DNA lesions are a direct risk to genome stability observed in t-MN [26]. Recent studies performed using next-generation sequencing have identified additional gene mutations in IDH1, ASXL1, SRSF2, SF3B1, SETBP1, TP53 and KTM2A-r in childhood t-MN [27]. These mutations were tracked backwards in bone marrow samples before the t-MN occurrence demonstrated the clonal evolution in t-MN with some somatic mutations preceding cytotoxic treatment and favouring leukemic development. Therefore, these genes are under scrutiny to test possible associations with environmental factors in the pathogenesis of i-AML.
Accepting the minimal two-step model for AML, in which the disruptive effect of abnormal gene rearrangements within cell signalling pathways originates a clone, failure in DNA repair system and/or modulation of susceptibility genetic factor, particularly with continued exposure to genotoxic substances, have been the pathogenic ripple, which needs to be uncovered.
Overview of MLL recombinome in i-AML
KTM2A drives essential biological processes through its DNA bind domains either directly (through sequences enriched for AT rich or nonmethylated CpG) or indirectly (through sequence specific transcription factors such as E2Fs), providing interfaces for the assembly of multiprotein complexes and methylate histone H3 at lysine 4 [28]. Chromosomal translocations involving the human MLL, nowadays called KMT2A gene, were associated with a dismal outcome in ALL, while in AML, some reports have demonstrated that some KMTA2 partner gene displays either a good or intermediate prognosis [3, 10, 29]. The break points for rearrangements in KMT2A occur mainly in 8-kb area between exon 8 and 13 (breakpoint cluster region) and accounts for 5%–10% of all acute leukaemia. In AML, KMT2A-r is recurrently associated with 35%–50% of infants and 5%–10% of therapy-related cases. The identification of the partner gene is necessary to provide optimal treatment stratification at diagnosis and prognosis.
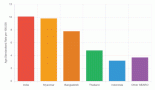
German investigators with a huge worldwide collaboration have created the MLL recombinome study that has been allowing us to identify new reciprocal translocations, complex rearrangements, internal duplications, deletions or inversions on the 11q23 region where the KMT2A is located and inserted into other chromosomes [30]. According to data from MLLrecombinome, the most frequent rearrangements occur either with MLLT3/AF9, MLLT1/ENL, ELL, MLLT10/AF10, MLLT4/AF6 or AFF1/AF4 genes, or are derived from gene internal duplications (MLL-PTDs) representing ~90% of AML cases. Amongst infants, breakpoints occur more frequently in KMT2A intron 11 [31]. However, breakpoint distribution for MLL-AF6 fusions displayed a clear preference for KMT2A intron 9 recombinations. Their occurrence differed significantly in the cohorts of infant, paediatric and adult leukaemia patients [30]. It is worthwhile to mention that the distribution of KMT2A-r in AML differs from ALL, either in infants, paediatric or adult patients, suggesting different biological mechanisms that may also reflect diverging factors. The extensive MLL recombinome analyses at the molecular level have allowed the identification of more than 71 different KMT2A-fusion partners [32]. To systematically classify the acute leukaemia with KMT2A-r, a multiplex RT-PCR (mRT-PCR) technique was standardised to identify the most frequent partners genes, such as MLLT1, MLLT3, MLLT4, MLLT10, AFF1 and the partial tandem duplication (MLL-PTD) [33]. In this context, the most common KMT2A fusion partners in our series of i-AML are: MLLT3/AF9 (35.7%), MLLT1/ENL (15.3%), MLLT4/AF6 (14.1%), MLLT10/AF10 (10%), AFF1/AF4 and MLL-PTD (6.1%). KMT2A with other partner genes or nonidentified are still underestimated because of the difficulty in their detection using only conventional cytogenetic technique, FISH and mRT-PCR. The KMT2A-r was morphologically more associated with AML-M4/M5, although was also identified in other AML subtypes, e.g., AML-M7. These groups altogether were associated with adverse prognosis [34].
The genome-wide technologies that demonstrated the genomic landscape of acute leukaemia may unravel the prenatal origin, throughout mutational signatures associated with environmental exposures.
Childhood AML associated with maternal pesticides exposures
Consensual evidence about risk factors and etiology of c-AML is still a challenge. It is well accepted that ionizing radiation exposures during gestation and genetic syndromes, e.g., DS, and familial monosomy 7 increase the risk of AML [35]. However, those factors account for only a small proportion of cases.
It is well known that children are more susceptible to environmental carcinogens than adults due to physiological vulnerability [36]. The immaturity of tissues, higher growing rates and cellular proliferation demand might jeopardise the haemopoiesis when the child is exposed to hazard substances [36]. Regarding this, exposures to pesticides through parental occupation or household chemicals have emerged as risk factors for childhood leukaemia and, particularly, c-AML [35].
Two meta-analyses showed that prenatal maternal occupational exposure to pesticides was strongly associated with c-AML. The risk estimates were (sOR: 2.64, 95% CI: 1.48–4.71) and (mRR 2.68; 95% CI 1.06–6.78) in each study [37, 38]. Indeed, a remarkable association amongst farm-related exposures and childhood leukaemia was observed (sOR: 2.44, 95% CI: 1.53–3.89) [37]. Those results were based on few studies that assessed risk associations for c-AML individually, e.g., without grouping with ALL within analysis.
A hospital-based case-control study from Children’s Cancer Study Group (CCG), which gathered 204 AML cases younger than 18 years and single-matched controls, showed a suggestive dose-dependent association of maternal occupational exposure to pesticides with c-AML risk in the offspring (p for trend = 0.008). Seven out of 204 c-AML had mothers exposed to pesticides for more than 1,000 days; for either parent exposed for more than 1,000 days, the association with c-AML was significant (OR: 3.8, 95% CI: 1.5–9.7) [39]. Another CCG study tested the risk association between paternal military service and childhood leukaemia in their offspring, considering the fact that during the 1980s, militaries were exposed to toxic substances, including 2, 3, 7, 8-tetracholodibenzo-p-dioxin used during the Vietnam War. A total of 605 c-AML and individually matched controls were included in the analysis, and results showed that c-AML risk was slightly increased among the offspring of veterans who had served in Vietnam (OR: 1.7, 95% CI: 1.0–2.9) [40]. More recently, a pooled analysis by the Childhood Leukaemia International Consortium (CLIC) gathered data from ten case-control studies, accruing 1,357 c-AML cases and 12,443 controls; in this study, c-AML risk was increased in the offspring of mothers who were occupationally exposed to pesticides during pregnancy (pooled OR: 1.94, 95% CI: 1.19–3.18) [41]. This group has also performed a meta-analysis including CLIC studies and other data published world-wide, and found that maternal occupational exposure to pesticide during pregnancy was associated with c-AML risk in the offspring (sOR: 3.30, 95% CI: 2.15–5.06) [41].
Residential exposure to pesticides during pregnancy was also associated with increased risk to childhood leukaemia, considering unspecified pesticides (sOR: 1.54, 95% CI: 1.13–2.11), insecticides (sOR: 2.05, 95% CI: 1.80–2.32) and herbicides (sOR: 1.61, 95% CI: 1.20–2.16). Specifically, for c-AML, an increased risk association was observed with insecticides (sOR: 1.85, 95% CI: 1.29–2.64) [42]. The same results were observed by the French group (the ESCALE Study) that a national registry-based case-control study gathered 100 c-AML and 1,681 controls. Significant associations were found between c-AML risk and maternal household use of any pesticides (OR: 2.2, 95% CI: 1.4–3.3) and insecticides (OR: 2.1, 95% CI: 1.4–3.3) [43]. More recently, the CLIC group reviewed 740 c-AML and 10,847 controls to test the time-points of maternal exposures. The risk for c-AML in the offspring was increased with maternal pesticide exposure at home within 1–3 months before conception (OR: 1.49, 95% CI: 1.02–2.16) and during pregnancy (OR: 1.21, 95% CI: 1.21–1.99) [44]. Boys were more affected by c-AML than girls with maternal exposure within 1–3 months before conception (OR: 1.77, 95% CI: 1.06–2.98) and during pregnancy (OR: 1.72, 95% CI: 1.22–2.43), as well as for c-AML diagnosed until four years with exposure to pesticides during pregnancy (OR: 2.08, 95% CI: 1.44–3.02). The main types of pesticides associated with c-AML risk were insecticides (OR: 1.55, 95% CI: 1.21–2.00) and pesticides used on pets (OR: 1.51, 95% CI: 1.07–2.12) [44].
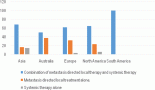
Regarding i-AML, four observational studies demonstrated associations of infant leukaemia with maternal exposure to pesticide during pregnancy, leading to the speculation of transplacental foetal exposure with DNA damage and chromosomal translocation as early pre-leukemic events [45–48]. The paucity of adequately powered studies is due to very low incidence of infant leukaemia. Only through collaborative studies is it possible to draw consistent conclusions. The first international collaborative study on infant leukaemia was conducted by Alexander et al that gathered patients with different costumes and genetic backgrounds (European, Middle East, South America and Asia countries) and accrued 74 i-AML cases (29 with KMT2A-r identified); they have shown that maternal pesticide exposure during pregnancy was associated with i-AML (OR: 5.08, 95% CI: 1.84–14.04) [45]. Then the Brazilian Collaborative Study Group of Infant Acute Leukaemia carried out a study taking into account the hypothesis of transplacental exposures with DNA damage. Sixty-two i-AML were recruited, excluding DS children, and pesticide exposures were analysed and reported since the first trimester of pregnancy until breastfeeding periods. Results have shown that maternal exposure to pesticides during pregnancy was associated with risk for overall infant leukaemia in crude analysis (OR: 2.23, 95% CI: 1.58–3.16); then, when the analysis was performed for i-AML, adjusted for sex, income, maternal age and birth weight, the risk association was (OR: 3.50 95% CI: 0.01–6.11) [46, 48].
Children exposure to pesticides occurs in different ways, from foetal period to late childhood, either through contamination of their parents’ work clothes or direct household residues in water, air, soil and food [42, 49]. Newborns are exposed across the placenta and through breastfeeding. The broad term of pesticide exposure might mislead conclusions because of the broad term, very well pointed out by Hernández and Menéndez [50]. In recent epidemiological studies, the household pesticides have been exploring separately [47, 48]. The Children’s Oncology Group within the 172 i-AML cohort showed no significant associations between household pesticide exposures, including insecticides, moth control, rodenticides, flea or tick control, herbicides, insect repellants and professional pest exterminations as variables and risk for infant leukaemia [47]. However, in Brazilian studies, maternal exposure to pesticide was associated with risk for i-AML diagnosed until 11 months of age (adjusted OR: 5.01, 95% CI: 1.97–12.7). The maternal exposure to pyrethroid (aOR: 3.39, 95% CI: 1.72–16.78) or organophosphates (OP) (aOR: 5.50, 95% CI: 1.44–21.03) was both associated with an increased risk for i-AML [48]. In a small study of infants born in an agricultural region in the Philippines, the prevalence of AML translocation with RUNX1-RUNXT1 in cord blood samples was about 2-fold higher among those with detectable meconium levels of the methylcarbamate insecticide propoxur [51]. Another interesting study is the association found between solvents exposure and AML previously analysed in a case-study carried out among adults in Italy. An association of the disease in individuals with RAS mutations and antecedents of exposure to solvents was observed (OR: 4.8, 95% CI: 1.2–18.8) [52, 53]. This finding is very interesting, because RAS mutation genes have been observed with variable frequencies (~15%–20%) in AML [54] and RAS mutations in early-age leukaemia were modulated by NQO1 rs1800566 (C609T), emphasizing the critical role of genetic susceptibility with somatic mutations in the mechanistic pathway leading to leukaemia in childhood [55].
The applications of various pesticides in agricultural and public health programs have caused severe environmental pollution and health hazards. It is a challenge to evaluate the exposure of individual substances (qualitative studies), although, Sala et al have measured the internal dose levels of organochlorines in paired settings of dyads, mother blood and cord blood samples from a rural village (Flix, Catalonia and Spain) located in the vicinity of an organochlorine-compound factory plant. All newborns presented detectable levels of organochlorine compounds, with higher values in samples from Flix than nearby villages [56]. This study demonstrated that the dissemination of pesticides compounds can exercise effects on nontarget organisms.
Brazil has been the world’s top pesticide market consumer as very well described by Albuquerque et al [57]. Therefore, epidemiological and experimental research must be performed in order to increase the level of information on the role of pesticides in i-AML.
Speculations of possible mechanisms how pesticides compounds drive leukemogenesis
Pesticides are chemical compounds that include insecticides (organochlorines, OP, carbamates and pyrethroids), herbicides (paraquat, diquat and 2,4-dichlorophenoxyacetic acid), fungicides (dithiocarbamates and captan), fumigants (ethylene dibromide and methyl bromide), rodenticides (anticoagulants to control rodents), and algicides, among others. Since the class of organochlorine came in restricted use, the second line of pesticides, i.e., OP and pyrethroids, became the most common group available [58]. For instance, the OP insecticide chlorpyrifos (O,O-diethyl O-3,5,6-trichloro-2-pyridyl phosphorothioate) has been widely used in indoor pest control (mainly for cockroaches and termites). Some in vitro studies (human cell lines) have demonstrated the genotoxic potential of OP inducing DSB and hypermethylation in CDKN1A [59]. The genotoxity of fungicides with the concentration of dose-response to Topo-II inhibition and induction of DSB was demonstrated in fruit flies [60]. It has been demonstrated that chlorpyrifos induces KMT2A-r through caspase 3-pathway with genomic instability and Topo-II inhibition in human foetal liver haematopoietic CD34+ cells [61].
It has been shown that pesticides may induce oxidative stress by producing free radicals, enhancing lipid peroxidation, causing a drastic activity of antioxidant enzymes in mammalian systems [62]. Moreover, these pesticides producing oxidative stress lead to toxicity in in vitro experiments (animal studies), and are found also in pesticide’s manufacturing workers and sprayers [63, 64]. Thus, DNA damage and oxidative stress are proposed mechanisms that link pesticide compounds to human pathogenesis of diseases observed in epidemiological studies [65, 66]. Long-lasting or acute oxidative stress disturbs cell metabolism. The reactive oxygen species (ROS) are able to produce permanent changes in the structure of proteins, lipids and DNA. The proteins that are oxidised may lose or enhance their activity. Moreover, the proteins oxidised are able to form aggregates that inhibit the systems responsible for protein degradation and lead to alterations of proteins in the cell [67].
Pesticide structures may be very stable, which means that they do not break down easily and can remain in the environment long after application and in organisms long after exposure. Some of these compounds present with slow degradation and subsequent bioaccumulation. In summary, the biological evidence linking pesticide compounds suggest that drugs interfering with DNA remodelling by Topo-II can mediate the formation of specific chromosomal translocation breakpoints in a similar pathway as chemotherapy agents.
Early-age AML, genetic susceptibility and genes–environment interactions
Genetic susceptibility has emerged as an important risk factor for childhood leukaemia, mainly because gene variants have modulated the attributable risk between environmental exposures and diseases. GWS have been providing lists of gene mutations, single nucleotide variations (SNP), expression frequencies and copy number variation (CNV) of candidate genes as potential biomarkers for genomic instability and novel therapeutic targets for children and adult cancer [68, 69]. However, very few studies have been conducted underlying genetic susceptibility in early-age AML with GWAS approaches.
Genetic syndromes are well established factors associated with high predisposition for childhood leukaemia and it accounts for about 10% of cases [35, 70]. A sum of multiples, independent or complementary genetic lesions are required for the development of a malignant disease from a haematopoietic progenitor clone. Maternal and infant diet, smoking, pesticides, household chemicals, automobile smoke and paint are the most important environmental risk factors for childhood leukaemia. Thus, polymorphisms in genes related to the metabolism of procarcinogenic substances may increase the risk of developing the disease. With the rationale of t-MN, the profile of genomic aberrations found in i-AML as well as the case-control studies, we performed investigations in several genes along the xenobiotic, gene variants in the base-excision repair (BER) and nonhomolog junction repair (n-HJR) systems. The enzymes of the xenobiotic system are capable of directly influencing the activation or inactivation of these compounds in the body. SNPs in gene involved with immune system pathways, cell cycle, DNA repair, folate metabolism and methylome process are also investigated in risk susceptibility to childhood leukaemia [71].
The equilibrium between the activities of Phase I and II enzymes of the xenobiotic system is essential for the organism’s response to environmental insults to haemostasis, efficiency and maintenance of human genetic material. A systematic review that included the great majority of the studies carried out in this topic has identified about 50 studies that found consistent results with the estimated rates of genetic susceptibility and risk of pediatric leukaemia. The polymorphisms most frequently investigated were those located in the genes of the cytochrome P450 family (CYPs), GSTM1, GSTT1 and NQO1 [72]. Some studies have shown that certain haplotypes of CYP1A1 are associated with an increased risk for paediatric leukaemia (ALL and AML) whose parents are smokers (OR: 2.1) [73], while CYP2E1* 5 allele in heterozygosity was associated with the risk of AML in children in Turkey (OR: 4.9) [74]. The GSTM1 and GSTT1 genes encode for glutathione S-transferases μ1 and θ1, respectively, which function in detoxifying processes of electrophilic xenobiotics, such as chemical carcinogens, and environmental pollutants, and inactivate secondary metabolites from endogenous oxidative stress. GSTM1 and GSTT1 alleles can both be deleted (null genotypes), which result in the absence of the protein [75–77]. In our hands, the GSTM1/T1 null genotype conferred increased risks to AML (aOR: 2.14).
The genetic variant NQO1 609C > T (rs1800566) known as NQO1*2 allele encodes a nonsynonymous mutation (Pro187Ser) that confers the lack of enzyme activity, and has been associated with the increased risk for benzene poisoning among benzene-exposed workers (7.6-fold rate) [78]. The NQO1gene encodes for a cytosolic flavoenzyme, NAD(P)H dehydrogenase quinone 1, that catalyses the two-electron reduction of quinones to hydroquinones, preventing the formation of ROS generated by redox cycling of semiquinones, thus functioning as a detoxifying enzyme [79].
The detoxification enzyme NAD(P)H: quinoneoxidoreductase (NQO1) is a flavoenzyme that detoxifies benzene metabolites, quinines and azo-dyes. NQO1 has an important function to protect cells against mutagenecity from free radicals and oxygen metabolites [79]. The NQO1 C609T (P187S) polymorphisms have been strongly associated with the risk of childhood ALL particularly for IL with KMT2A-r [80]. We have found that NQO1 C609T modified risk in pediatrics leukaemia depending on age range and the variant genotype in combinations with other gene polymorphisms, such as Paraoxonase 1 (PON1) gene variant [75]. PON1 functions are to oxidize and hydrolyse metabolites of several organophosphorus pesticides. Its functional activity is very important in the cellular oxidative stress process [81]. The most common PON1 polymorphisms A21439G (PON1 Q192R) and T12801A (PON1 L55M) cause variability in enzyme activity, affecting its sensitivity to xenobiotic metabolism in young individuals [82]. We have investigated NQO1 and PON1 polymorphisms associated with early-age leukaemia considering acquiring genomic aberrations and age at the onset of the disease. We have found that infant leukaemia with KTM2A-r was strongly associated with NQO1 C609T variant genotypes (OR: 2.93), while PON1L55M polymorphism increased the risk of ALL in children aged ≥13 months (OR: 3.2) [75].
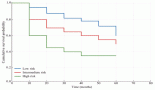
Genetic polymorphisms conferred by SNPs in the DNA repair genes pathways have also been associated with genetic susceptibility to paediatric leukaemia. For instance, a Taiwanian study, which included 266 cases of paediatric leukaemia, found that XRCC4 (x-ray repair cross complementing 4) haplotypes, composed of SNPs rs6869366 and rs28360071, were associated with the increased risk for the disease, without distinguishing subtypes [83]. There is as yet no evidence in the literature regarding genetic susceptibility to AML specifically. We are performing genotyping studies with the aim to identify gene variants in BER and n-HJR systems in order to estimate the associations with i-AML/KTM2A positive. BER is crucial in the elimination process of single-strand damages with reinsertion of band religation. In homeostasis, the XRCC1 is responsible for recruitment and anchoring ligase-enzymes during the double-strand religation. So far, XRCC1 399A > G confers an increased risk for i-AML as whole. In addition, in our investigation, the DIP3-XRCC4 was found associated with infants KTM2A-r (OR: 3.2). In a model system to investigate the function of XRCC genes, the DIP3 (ins/del, 30pb of intron3) of XRCC4 had substantially decreased the XRCC4 protein levels leading to reduced cellular ligase IV activity in vitro and in vivo studies.
Conclusion
In this review, we have revisited early-age AML, a group that comprises distinct AML with KTM2A-r and is associated with transplacental exposures. We have comprehensively summarised the epidemiologic and biological evidence that sustains the hypothesis of leukaemogenesis initiating upon transplacental maternal exposures. Few observational studies have estimated the risk association of i-AML with maternal exposures during pregnancy to pesticide compounds. We have put together the results of the Brazilian studies and the review of mechanistic investigations that support the biological plausibility of observational findings. The genomewide technologies that demonstrated the genomic landscape of acute leukaemia might unravel the prenatal origin throughout mutational signatures, and the challenge is to combine cell biology associated with environmental exposures. Thus, future work should explore the associations of genes involved in cell detoxification concerning the myeloid compartment and target for AML.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Acknowledgments
The authors thank the physicians section of the Brazilian Collaborative Study Group of Infant Acute Leukemia (BCSGIAL) from different Brazilian regions who sent samples and clinical data for i-AML investigations. MSPO has CNPq scholarship (#301594/2015) and the Infant Leukemia Project had SwissBridge grant support.
References
1. Howlader N et al (2012)SEER Cancer Statistic Review, 1975-2009 Vintage 2009 Populations (Bethesda: National Cancer Institute)
2. Kelly LM and Gilliland DG (2002) Genetics of myeloid leukemias Annu Rev Genomics Hum Genet 3 179–98 https://doi.org/10.1146/annurev.genom.3.032802.115046 PMID: 12194988
3. Balgobind BV, Raimondi SC, and Harbott J, et al (2009) Novel prognostic subgroups in childhood 11q23/MLL-rearranged acute myeloid leukaemia: results of an international retrospective study Blood 114 2489–2496 https://doi.org/10.1182/blood-2009-04-215152 PMID: 19528532 PMCID: 2927031
4. Greaves MF and Wiemels J (2003) Origins of chromosome translocations in childhood leukaemia Nat Rev Cancer 3 639–649 https://doi.org/10.1038/nrc1164 PMID: 12951583
5. Zwaan CM, Kolb EA, and Reinhardt D, et al (2015) Collaborative efforts driving progress in pediatric acute myeloid leukaemia J Clin Oncol 33(27) 2949–2962 https://doi.org/10.1200/JCO.2015.62.8289 PMID: 26304895 PMCID: 4567700
6. Andrade FG, Noronha EP, and Brisson GD, et al (2016) Molecular characterization of pediatric acute myeloid leukaemia: results of a multicentric study in Brazil Archives of Medical Research, in press. https://doi.org/10.1016/j.arcmed.2016.11.015
7. Tosi S, Harbott J, and Teigler-Schlegel A, et al (2000) t(7;12)(q36;p13), a new recurrent translocation involving ETV6 in infant leukaemia Genes, Chromosom, & Cancer 29 325–332
8. Carroll A, Civin C, and Schneider N, et al (1991) The t(1;22)(p13;q13) is a nonrandom and restricted to infants with acute megakaryoblastic leukaemia: a pediatric oncology group study Blood 78 748–752 PMID: 1859887
9. Gruber TA, Gedman AL, and Zhang J, et al (2012) An inv(16)(p13.3q24.3)-encoded CBFA2T3-GLIS2 fusion protein defines an aggressive subtype of pediatric acute megakaryoblasticleukaemia Cancer Cell 22 683–97 https://doi.org/10.1016/j.ccr.2012.10.007 PMID: 23153540 PMCID: 3547667
10. Masetti R (2014) Infants with acute myeloid leukaemia treated according to the Associazione Italiana di Ematologia e Oncologia Pediatrica 2002/01 protocol have an outcome comparable to that of older children Haematologica 99(8) 127–129 https://doi.org/10.3324/haematol.2014.106526
11. Andrade FG, Noronha EP, and Baseggio RM, et al (2016) Identification of the MYST3-CREBBP fusion gene in infants with acute myeloid leukaemia and hemophagocytosis Rev Bras Hematol Hemoter 38(4) 291–297 https://doi.org/10.1016/j.bjhh.2016.06.005 PMID: 27863755 PMCID: 5119666
12. Zweidler-McKay PA and Hilden JM (2008) The ABCs of infant leukaemia CurrProblPediatrAdolesc Health Care 38(3) 78–94
13. Loh ML (2011) Recent advances in the pathogenesis and treatment of juvenile myelomonocytic leukaemia Br J Haematol 152(6) 677–687 https://doi.org/10.1111/j.1365-2141.2010.08525.x PMID: 21623760
14. Wechsler J, Greene M, and McDevitt MA, et al (2002) Acquired mutations in GATA-1 in the megakaryoblastic leukaemia of Down syndrome Nat Genet 32 148–152 https://doi.org/10.1038/ng955 PMID: 12172547
15. Klusmann JH, Godinho FJ, and Heitmann K, et al (2010) Developmental stage-specific interplay of GATA-1 and IGF signaling in fetal megakaryopoiesis and leukemogenesis Genes Dev 24 1659–1672 https://doi.org/10.1101/gad.1903410 PMID: 20679399 PMCID: 2912563
16. Moura SV, Andrade F, and Magalhães IQ, et al (2015) Clinical and molecular epidemiology of neonatal leukaemia in Brazil Leuk Lymphoma 56(4) 903–909 https://doi.org/10.3109/10428194.2014.938327
17. Pine SR, Guo Q, and Yin C, et al (2007) Incidence and clinical implications of GATA-1 mutations in newborns with Down syndrome Blood 110 2128–2131 https://doi.org/10.1182/blood-2007-01-069542 PMID: 17576817
18. Al-Kasim F, Doyle JJ, and Massey GV, et al (2002) Incidence and treatment of potentially lethal disease in transient leukaemia of Down syndrome: Pediatric Oncology Group Study J Peditr Hematol Oncol 24 9–13 https://doi.org/10.1097/00043426-200201000-00004
19. Massey GV, Zipursky A, and Chang MN, et al (2006) A prospective study of the natural history of transient leukaemia (TL) in neonates with Down syndrome(DS):Children’s Oncology Group (COG) study POG-9481 Blood 107 4606–4613 https://doi.org/10.1182/blood-2005-06-2448 PMID: 16469874
20. Greaves MF, Maia AT, and Wiemels JL, et al (2003) Leukaemia in twins: lessons in natural history Blood 102(7) 2321–2333 https://doi.org/10.1182/blood-2002-12-3817 PMID: 12791663
21. Gale KB, Ford AM, and Repp R, et al (1997) Backtracking leukaemia to birth: identification of clonotypic gene fusion sequences in neonatal blood spots Proc Natl Acad Sci USA 94(25) 13950–13954 https://doi.org/10.1073/pnas.94.25.13950
22. Larson RA (2009) Therapy-related myeloid neoplasms Haematologica 94(4) 454–459 https://doi.org/10.3324/haematol.2008.005157 PMID: 19336749 PMCID: 2663607
23. Ranjbar A, Pasalar P, and Abdollahi M (2002) Induction of oxidative stress and acetylcholinesterase inhibition in organophosphorous pesticide manufacturing workers Hum Exp Toxicol 21(4) 179–182 https://doi.org/10.1191/0960327102ht238oa PMID: 12099619
24. Cho HW, Choi YB, and Yi ES, et al (2016) Therapy-related myeloid neoplasms in children and adolescents Blood Res 51(4) 242–248 https://doi.org/10.5045/br.2016.51.4.242
25. Gilliland DG, Jordan CT, and Felix CA (2004) The molecular basis of leukaemia Hematology 1 80–97 https://doi.org/10.1182/asheducation-2004.1.80
26. Reed Williams WL et al (2002) DNA topoisomerase II single-strand nick model of MLL translocations in de novo leukemias with t(4;11)(q21;q23) Proc AACR 43 684
27. Fabiani E, Falconi G, and Fianchi L, et al (2017) Clonal evolution in therapy-related neoplasms Oncotarget 8(7) 12031–12040 PMID: 28076841 PMCID: 5355323
28. Liu H, Cheng EH, and Hsieh JJ (2009) MLL fusions: pathways to leukaemia Cancer Biol Ther 8(13) 1204–1211 https://doi.org/10.4161/cbt.8.13.8924 PMID: 19729989 PMCID: 3289713
29. Tomizawa D (2015) Recent progress in the treatment of infant acute lymphoblastic leukaemia Pediatr Int 57(5) 811–819. https://doi.org/10.1111/ped.12758 PMID: 26215843
30. Meyer C, Hofmann J, and Burmeister T, et al (2013) The MLL recombinome of acute leukemias in 2013 Leukaemia 27(11) 2165–2176 https://doi.org/10.1038/leu.2013.135
31. Emerenciano M, Meyer C, and Mansur MB, et al (2013) The distribution of MLL breakpoints correlates with outcome in infant acute leukaemia Br J Haematol 161(2) 224–236 https://doi.org/10.1111/bjh.12250 PMID: 23432364
32. Marschalek R (2011) Mechanisms of leukemogenesis by MLL fusion proteins Br J Haematol 152(2) 141–154 https://doi.org/10.1111/j.1365-2141.2010.08459.x
33. Burmeister T, Meyer C, and Gröger D, et al (2015) Evidence-based RT-PCR methods for the detection of the 8 most common MLL aberrations in acute leukemias Leuk Res 39(2) 242–247 https://doi.org/10.1016/j.leukres.2014.11.017
34. Emerenciano M, Barbosa Tda C, and de Almeida Lopes B, et al (2015) Subclonality and prenatal origin of RAS mutations in KMT2A (MLL)-rearranged infant acute lymphoblastic leukaemia Br J Haematol 170(2) 268–271 https://doi.org/10.1111/bjh.13279 PMID: 25613690
35. Puumala SE, Ross JA, and Aplenc R, et al (2013) Epidemiology of childhood acute myeloid leukaemia Pediatr Blood Cancer 60(5) 728–733 https://doi.org/10.1002/pbc.24464 PMID: 23303597 PMCID: 3664189
36. Whyatt RM and Perera FP (1995) Application of biologic markers to studies of environmental risks in children and the developing fetus Environ Health Perspect 103 6105–6110. https://doi.org/10.1289/ehp.95103s6105
37. Wigle DT, Turner MC, and Krewski D (2009) A systematic review and meta-analysis of childhood leukaemia and parental occupational pesticide exposure Environ Health Perspect 117(10) 1505–1513 https://doi.org/10.1289/ehp.0900582 PMID: 20019898 PMCID: 2790502
38. Van Maele-Fabry G, Lantin AC, and Hoet P, et al (2010) Childhood leukaemia and parental occupational exposure to pesticides: a systematic review and meta-analysis Cancer Causes Control 21(6) 787–809 https://doi.org/10.1007/s10552-010-9516-7 PMID: 20467891
39. Buckley JD, Robison LL, and Swotinsky R, et al (1989) Occupational exposures of parents of children with acute nonlymphocytic leukaemia: a report from the Children’s Cancer Study Group Cancer Res 49(14) 4030–4037 PMID: 2736544
40. Wen WQ, Shu XO, and Steinbuch M, et al (2000) Paternal military service and risk for childhood leukaemia in offspring Am J Epidemiol 151(3) 231–240 https://doi.org/10.1093/oxfordjournals.aje.a010198 PMID: 10670547
41. Bailey HD, Fritschi L, and Infante-Rivard C, et al (2014) Parental occupational pesticide exposure and the risk of childhood leukaemia in the offspring: findings from the childhood leukaemia international consortium Int J Cancer 135(9) 2157–2172 https://doi.org/10.1002/ijc.28854 PMID: 24700406 PMCID: 4845098
42. Turner MC, Wigle DT, and Krewski D (2010) Residential pesticides and childhood leukaemia: a systematic review and meta-analysis Environ Health Perspect 118(1) 33–41 PMID: 20056585 PMCID: 2831964
43. Rudant J, Menegaux F, and Leverger G, et al (2007) Household exposure to pesticides and risk of childhood hematopoietic malignancies: the ESCALE study (SFCE) Environ Health Perspect 115(12) 1787–1793 https://doi.org/10.1289/ehp.10596 PMID: 18087601 PMCID: 2137105
44. Bailey HD, Infante-Rivard C, and Metayer C, et al (2015) Home pesticide exposures and risk of childhood leukaemia: findings from the childhood leukaemia international consortium Int J Cancer 2015 137(11) 2644–2663 https://doi.org/10.1002/ijc.29631 PMID: 26061779 PMCID: 4572913
45. Alexander FE, Patheal SL, and Biondi A, et al (2001) Transplacental chemical exposure and risk of infant leukaemia with MLL gene fusion Cancer Res 61(6) 2542–2546 PMID: 11289128
46. Pombo-de-Oliveira MS, Koifman S, and Brazilian Collaborative Study Group of Infant Acute Leukaemia (2006) Infant acute leukaemia and maternal exposures during pregnancy Cancer Epidemiol Biomarkers Prev 15(12) 2336–2341 https://doi.org/10.1158/1055-9965.EPI-06-0031 PMID: 17164354
47. Slater ME, Linabery AM, and Spector LG, et al (2011) Maternal exposure to household chemicals and risk of infant leukaemia: a report from the Children’s Oncology Group Cancer Causes Control 22(8) 1197–1204 https://doi.org/10.1007/s10552-011-9798-4 PMID: 21691732 PMCID: 4836386
48. Ferreira JD, Couto AC, and Pombo-de-Oliveira MS, et al (2013) In utero pesticide exposure and leukaemia in Brazilian children < 2 years of age Environ Health Perspect 121(2) 269–275 PMCID: 3569673
49. Rudant J, Clavel J, and Infant-Rivard C (2010) Selection bias in case-control studies in household exposure to pesticides and childhood acute leukaemia J Expo Sci Environ Epidemiol 20(4) 299–309 https://doi.org/10.1038/jes.2009.61
50. Hernández AF and Menéndez P (2016) Linking pesticide exposure with pediatric leukaemia: potential underlying mechanisms Int J Mol Sci 17(4) 461 https://doi.org/10.3390/ijms17040461
51. Lafiura KM, Bielawski DM, and Posecion NC, et al (2007) Association between prenatal pesticide exposures and the generation of leukaemia-associated t(8;21) Pediatr Blood Cancer 49(5) 624–628 https://doi.org/10.1002/pbc.21283 PMID: 17610268
52. Barletta E, Gorini G, and Vineis P, et al (2004) Ras gene mutations in patients with acute myeloid leukaemia and exposure to chemical agents Carcinogenesis 25(5) 749–755 https://doi.org/10.1093/carcin/bgh057
53. Algualcil J, Porta M, and Kauppinen T, et al (2003) Occupational exposure to dyes, metals, polycyclic aromatic hydrocarbons, and other agents and K-ras activation in human exocrine pancreatic cancer Int J Cancer 107(4) 635–641 https://doi.org/10.1002/ijc.11431
54. Liang DC, Shih LY, and Fu JF, et al (2006) K-RAS mutations and N-RAS mutations in childhood acute leukemias with or without mixed-linage leukaemia gene rearrangements Cancer 106(4) 950–956 https://doi.org/10.1002/cncr.21687 PMID: 16404744
55. Andrade FG Furtado-Silva JM, and de Aguiar Gonçalves BA, et al (2014) RAS mutations in early age leukaemia modulated by NQO1 rs1800566 (C609T) are associated with second-hand smoking exposures BMC Cancer 14 133 https://doi.org/10.1186/1471-2407-14-133 PMID: 24571676 PMCID: 3946262
56. Sala M, Ribas-Fitó N, and Cardo E, et al (2001) Levels of hexachlorobenzene and other organochlorine compounds in cord blood: exposure across placenta Chemosphere 43(4–7) 895–901 https://doi.org/10.1016/S0045-6535(00)00450-1 PMID: 11372882
57. Albuquerque AF, Ribeiro JS, and Kummrow F, et al (2016) Pesticides in Brazilian freshwaters: a critical review Environ Sci Process Impacts 18(7) 779–787 https://doi.org/10.1039/C6EM00268D PMID: 27367607
58. George J and Shukla Y (2011) Pesticides and cancer: insights into toxicoproteomic-based findings J Proteomics 74(12) 2713–2722 https://doi.org/10.1016/j.jprot.2011.09.024 PMID: 21989265
59. Williams RD, Boros LG, and Kolanko CJ, et al (2004) Chromosomal aberrations in human lymphocytes exposed to the anticholinesterase pesticide isofenphos with mechanisms of leukemogenesis Leuk Res 28(9) 947–958 https://doi.org/10.1016/j.leukres.2003.12.014 PMID: 15234572
60. Rahden-Staron I (2002) The inhibitory effect of the fungicides captan and captafol on eukaryotic topoisomerases in vitro and lack of recombinagenic activity in the wing spot test of Drosophila melanogaster Mutat Res 518(2) 205–213 https://doi.org/10.1016/S1383-5718(02)00107-9 PMID: 12113771
61. Lu C, Liu X, and Liu C, et al (2015) Chlorpyrifos induces MLL translocations through caspase 3-dependent genomic instability and topoisomerase II inhibition in human fetal liver hematopoietic stem cells Toxicol Sci 147(2) 588–606 https://doi.org/10.1093/toxsci/kfv153 PMID: 26198043
62. George J and Shukla Y (2011) Pesticides and cancer: insights into toxico proteomic-based findings J Proteomics 74(12) 2713–2722 https://doi.org/10.1016/j.jprot.2011.09.024 PMID: 21989265
63. Gultekin F, Delibas N, and Yasar S, et al (2001) In vivo changes in antioxidant systems and protective role of melatonin and a combination of vitamin C and vitamin E on oxidative damage in erythrocytes induced by chlorpyrifos-ethyl in rats Arch Toxicol 75 88–96 https://doi.org/10.1007/s002040100219 PMID: 11354911
64. Lopez O, Hernández AF, and Rodrigo L, et al (2007) Changes in antioxidant enzymes in humans with long-term exposure to pesticides Toxicol Lett 171 146–153 https://doi.org/10.1016/j.toxlet.2007.05.004 PMID: 17590542
65. Olinski R, Gackowski D, and Foksinski M, et al (2002) Oxidative DNA damage: assessment of the role in carcinogenesis, atherosclerosis, and acquired immunodeficiency syndrome Free Rad Biol Med 33 192–200 https://doi.org/10.1016/S0891-5849(02)00878-X PMID: 12106815
66. Muniz JF, McCauley L, and Scherer J, et al (2008) Biomarkers of oxidative stress and DNA damage in agriculturalworkers: a pilot study ToxicolAppl Pharmacol 227 97–107
67. Tvrdá E1, Kňažická Z, and Bárdos L, et al (2011) Impact of oxidative stresson male fertility: a review Acta Vet Hung 59 465–484 https://doi.org/10.1556/AVet.2011.034
68. Chae YK, Anker JF, and Carneiro BA, et al (2016) Genomic landscape of DNA repair genes in cancer Oncotarget 7(17) 23312–23321 https://doi.org/10.18632/oncotarget.8196 PMID: 27004405 PMCID: 5029628
69. Farrar JE, Schuback HL, and Ries RE, et al (2016) Genomic profiling of pediatric acute myeloid leukaemia reveals a changing mutational landscape from disease diagnosis to relapse Cancer Res 76(8) 2197–2205 https://doi.org/10.1158/0008-5472.CAN-15-1015 PMID: 26941285 PMCID: 4873364
70. Deschler B and Lubbert M (2006)Acute myeloid leukaemia: epidemiology and etiology Cancer 107 2099–2107 https://doi.org/10.1002/cncr.22233 PMID: 17019734
71. Chokkalingam AP and Buffler PA (2008) Genetic susceptibility to childhood leukaemia Radiat Prot Dosimetry 132(2) 119–129 https://doi.org/10.1093/rpd/ncn255 PMID: 18922824 PMCID: 2879095
72. Brisson GD, Alves LR, and Pombo-de-Oliveira MS (2015) Genetic susceptibility in childhood acute leukaemias: a systematic review Ecancermedicalscience 9 539 https://doi.org/10.3332/ecancer.2015.539 PMID: 26045716 PMCID: 4448992
73. Lee KM, Ward MH, and Han S, et al (2009) Paternal smoking, genetic polymorphisms in CYP1A1 and childhood leukaemia risk Leukaemia Res 33 250–258 https://doi.org/10.1016/j.leukres.2008.06.031
74. Aydin-Sayitoglu M, Hatirnaz O, and Erensoy N, et al (2006) Role of CYP2D6, CYP1A1, CYP2E1, GSTT1, and GSTM1 genes in the susceptibility to acute leukemias. Am J Hematol 81(3) 162–70 https://doi.org/10.1002/ajh.20434 PMID: 16493615
75. De AguiarGonçalves BA, Vasconcelos GM, and Thuler LCS, et al (2012) NQO1 rs1800566 (C609T), PON1 rs662 (Q192R), and PON1 rs854560 (L55M) polymorphisms segregate the risk of childhood acute leukemias according to age range distribution Cancer Causes Control 23(11) 1811–1819 https://doi.org/10.1007/s10552-012-0060-5
76. Zanrosso CW et al (2012) Genetic variability in N-acetyltransferase 2 gene determines susceptibility to childhood lymphoid or myeloid leukaemia in Brazil Leuk Lymphoma 53(2) 323–327 https://doi.org/10.3109/10428194.2011.619605
77. Lopes BA, Emerenciano M, and Gonçalves BAA, et al (2015) Polymorphisms in CYP1B1, CYP3A5, GSTT1, and SULT1A1 are associated with early age acute leukaemia PLoS One 10(5) e0127308 https://doi.org/10.1371/journal.pone.0127308 PMCID: 4436276
78. Rothman N, Smith MT, and Hayes RB, et al (1997) Benzene poisoning, a risk factor for hematological malignancy, is associated with the NQO1 609C-->T mutation and rapid fractional excretion of chlorzoxazone Cancer Res 57(14) 2839–2842 PMID: 9230185
79. Vasiliou V, Ross D and Nebert DW (2006) Update of the NAD(P)H:quinoneoxidoreductase (NQO) gene family Hum Genomics 2(5) 329–335 https://doi.org/10.1186/1479-7364-2-5-329 PMID: 16595077 PMCID: 3500182
80. Smith MT, Wang Y, and Skibola CF, et al (2002) Low NAD(P)H:quinoneoxidoreductase activity is associated with increased risk of leukaemia with MLL translocations in infants and children Blood 100 4590–4593 https://doi.org/10.1182/blood-2001-12-0264 PMID: 12393620
81. Costa LG, Vitalone A, Cole TB, et al (2005) Modulation of paraoxonase (PON1) activity Biochem Pharmacol 69 541–550 https://doi.org/10.1016/j.bcp.2004.08.027 PMID: 15670573
82. Huen K, Harley K, and Brooks J, et al (2009) Developmental changes in PON1 enzyme activity in young children and effects of PON1 polymorphisms Environ Health Perspect 117 1632–1638 https://doi.org/10.1289/ehp.0900870 PMID: 20019917 PMCID: 2790521
83. Wu KH, Wang CH, and Yang YL, et al (2010) Significant association of XRCC4 single nucleotide polymorphisms with childhood leukaemia in Taiwan Anticancer Research 30 529–534