Functional pancreatic neuroendocrine tumour causing Cushing’s syndrome: the effect of chemotherapy on clinical symptoms
Paulo Henrique do Amor Divino1, Katia Regina Marchetti1, Madson Q Almeida1 and Rachel P Riechelmann2
1Instituto do Câncer do Estado de São Paulo, Av Dr Arnaldo, 251 - Cerqueira César, Sao Paulo, 1246-000, Brazil
2AC Camargo Cancer Center, Sao Paulo 01509-900, Brazil
Correspondence to: Rachel P Riechelmann and Paulo Henrique Amor Divino. Email: rachelri2005@gmail.com and phadmed@hotmail.com
Abstract
Background: Neuroendocrine tumours (NETs) are a heterogeneous group of diseases that can originate from any part of the gastrointestinal tract, bronchi, thyroid and pancreas. These tumours may be functioning or not depending on their ability to produce active substances, such as adrenocorticotrophic hormone (ACTH). ACTH-producing pancreatic neuroendocrine tumours are rare, with limited data about effective antitumor therapies.
Case Report: A 58-year-old man with a history of type-2 diabetes mellitus and arterial hypertension was diagnosed with Cushing’s syndrome (CS) secondary to an ACTH ectopic production from a well-differentiated neuroendocrine tumour of the pancreas metastatic to the liver. The patient underwent initial body-caudal pancreatectomy, splenectomy and hepatic nodulectomy with subsequent recurrence. Hepatic embolisation and somatostatin analogues were used to control CS but without success. Bilateral adrenalectomy led to CS control, while capecitabine and oxaliplatin (CAPOX) was effective in controlling tumour growth and ACTH production.
Discussion: ACTH-producing pancreatic neuroendocrine tumours are rare, aggressive and difficult to treat with available therapies. In settings of limited resources, such as in developing countries where targeted therapies are not available, cytotoxic chemotherapy with CAPOX represents a good and inexpensive option to control ACTH-producing pancreatic neuroendocrine tumours. Because of its complexity, the management of this tumour should be performed by multidisciplinary teams.
Keywords: neuroendocrine tumours, Cushing syndrome, pancreatic neoplasms
Copyright: © the authors; licensee ecancermedicalscience. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Published: 13/10/2017; Received: 02/05/2017
Introduction
Neuroendocrine tumours (NETs) are a heterogeneous group of diseases that can originate in any part of the gastrointestinal tract, bronchi, thyroid and pancreas [1]. They are rare diseases and probably underdiagnosed, since in many cases the course of the disease is indolent. Functioning pancreatic NETs (PNETs) are rare, representing approximately 1% of pancreatic neoplasias [2, 3]. Among these tumours, those that produce adrenocorticotrophic hormone (ACTH) are even rarer, with few reports in the literature about how to treat them [4]. Here, we report the case of 58-year-old male who developed Cushing’s syndrome (CS) secondary to ACTH production by a metastatic PNET and which was successfully controlled by a cytotoxic chemotherapy available in a setting of limited resources.
Case report
In December 2013, a 58-year-old man with a history of type-2 diabetes mellitus (DM2), systemic arterial hypertension (SAH) and dyslipidemia was referred to our centre, the Hospital de Clínicas de Sao Paulo, a large academic and public centre located in Sao Paulo, Brazil, from the primary care service to investigate uncontrolled blood pressure and blood glucose. At initial clinical examination, he presented facial phletora, hirsutism, violaceous striae and centripetal obesity, and a CS clinical diagnosis was suspected. Initial examinations identified hypercortisolism and elevation of ACTH (Table 1). The ACTH increment in the corticotropin-releasing hormone (CRH) stimulation test was 14%, suggesting an ectopic ACTH-producing tumour. To confirm this hypothesis, the patient underwent an inferior petrosal sinus sampling with CRH stimulation. The central/peripheral ACTH gradient was less than 2 at 0, 3, 5 and 10 min after desmopressin injection, confirming the ectopic ACTH producing. Magnetic resonance image (MRI) of the abdomen showed an expansive lesion in the tail of the pancreas measuring 5.0 cm in the largest diameter, multiple small nodules in hepatic segment III and increased volume of both adrenals (Figure 1). In January 2014, he was referred to our institution, the Instituto do Cancer do Estado de Sao Paulo, where he underwent an R0 body-caudal pancreatectomy, splenectomy and removal of the segment III of the liver, which was compatible with PNET, infiltrating the liver tissue. The final diagnosis was of a well-differentiated PNET, with positive immunohistochemistry staining of synaptophysin and chromogranin A, three mitosis/10 high-power fields and Ki67 index of 6%. There was adequate clinical control of DM2 and SAH after resection. In July 2014, a Gallium-68-DOTATATE PET-CT was performed and did not show any measurable metastatic disease. However, in November 2014, he presented hyperglycaemia and high blood pressure again, associated with elevation of both serum cortisol and ACTH, which were resultant from hepatic recurrence with a new lesion of 1.2 cm in segment VI of the liver. Because the patient was quite sick to undergo new hepatic resection due to uncontrolled CS and also because of the short interval from last metastasectomy, new surgical resection was contraindicated. Octreotide LAR 20 mg IM once every 28 days was started but was unsuccessful in controlling symptoms. Subsequent hepatic embolisation did not improve his condition either. Due to uncontrolled CS in February 2015, bilateral adrenalectomy was performed and the CS finally resolved. The pathology report revealed a metastatic neuroendocrine tumour in the left adrenal, with immunohistochemistry staining positive for synaptophysin, chromogranin A positive and ACTH, negative staining for CD56, CDX2 and TTF1 negative and ki67 index of 30%.
After symptom control, the patient was lost to follow up, returning six months with recurrent hyperglycaemia and skin hyperpigmentation; at that time, the elevation of plasma ACTH was identified. A new MRI of the abdomen showed a progression of hepatic metastases. Octreotide LAR was tried again, but there was disease progression after two doses. In October 2015, he received the combination of oxaliplatin 130 mg/m2 given on day one and capecitabine 1000 mg/m2 (CapOx) orally for 14 days, in a 21-days cycle. At this time, he presented diffuse exuberant hyperpigmented lesions of the skin, mainly in interphalangeal joints and tongue (Figure 2). After two cycles, there was significant improvement in cutaneous hyperpigmentation and the patient was restaged with new CT scans that showed stable disease. Despite good tolerance, the patient requested to stop chemotherapy and he went on chemoholiday. In March 2016, after three months without treatment, the hyperpigmented lesions of the skin worsened and imaging tests evidenced new progression of liver disease. Re-exposure to CapOx was indicated. He received three more cycles, when in in April/2016, CT scans demonstrated partial response in liver lesions. The patient chose to pause the chemotherapy once again. At the last image evaluation, in June 2017, the tumour has remained stabilised and three has been no further worsening of skin hyperpigmentation.
Table 1. Laboratory tests performed during treatment.
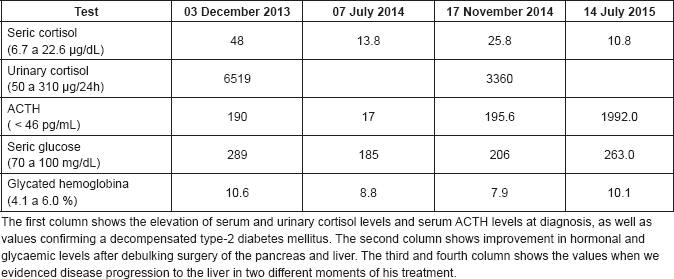
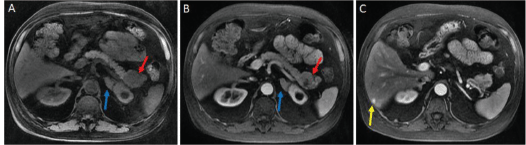
Figure 1. A: magnetic resonance imaging performed in the non-contrasting T1 sequence, showing an enlarged and irregular expansive lesion of the pancreas tail (red arrow) and thickening of the left adrenal (blue arrow). B: magnetic resonance imaging performed in the contrasting T1 sequence. C: nodular lesion in segment VI of the liver (yellow arrow).
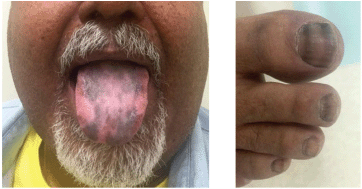
Figure 2. Hyperpigmented lesions of his tongue and nails likely caused by increased propiomelanocortin (POMC) prohormone production, which is cleaved into ACTH and MSH, inducing darkening of the skin and nails. The strips in his nails timely occurred when the tumour responded to CApOx
Discussion
While the most frequent functional PNETs are insulinoma and gastrinoma, ectopic adrenocorticotropic hormone-producing tumours are very rare types of PNET [5, 6] and carry a poor prognosis. The survival analysis of all the cases reported in the English/Spanish literature found that only 35% of patients were alive at 5 years after diagnosis [7]. Maragliano et al have reported a series of 124 cases of ACTH-secreting PNETs, in which female patients predominated (66%), the mean age at diagnosis was 42.1 years, 45% arose from the tail of the pancreas and most tumours were well differentiated (94%) [7]. Up to 78.7% of ACTH-secreting PNETs have exhibited distant metastases when diagnosed, with the most common sites being liver and lungs followed by the adrenal glands, bone, kidney and omentum [8].
The ectopic ACTH syndrome is responsible for 10–15% of all causes of CS. The symptoms intensity depends on the cortisol serum levels and the tumour aggressiveness. The clinical manifestations are diverse, from incidental diagnosis in asymptomatic patients to classical CS features, with proximal muscle fatigue, striae, skin pigmentation, hypertension, abdominal pain and high susceptibility to infection. The most frequent tumours associated with ectopic production of ACTH are small-cell carcinoma of the lung, followed by pancreatic, bronchial carcinoid tumours, thymic carcinoid tumours, medullary thyroid carcinoma and pheochromocytoma [9–11].
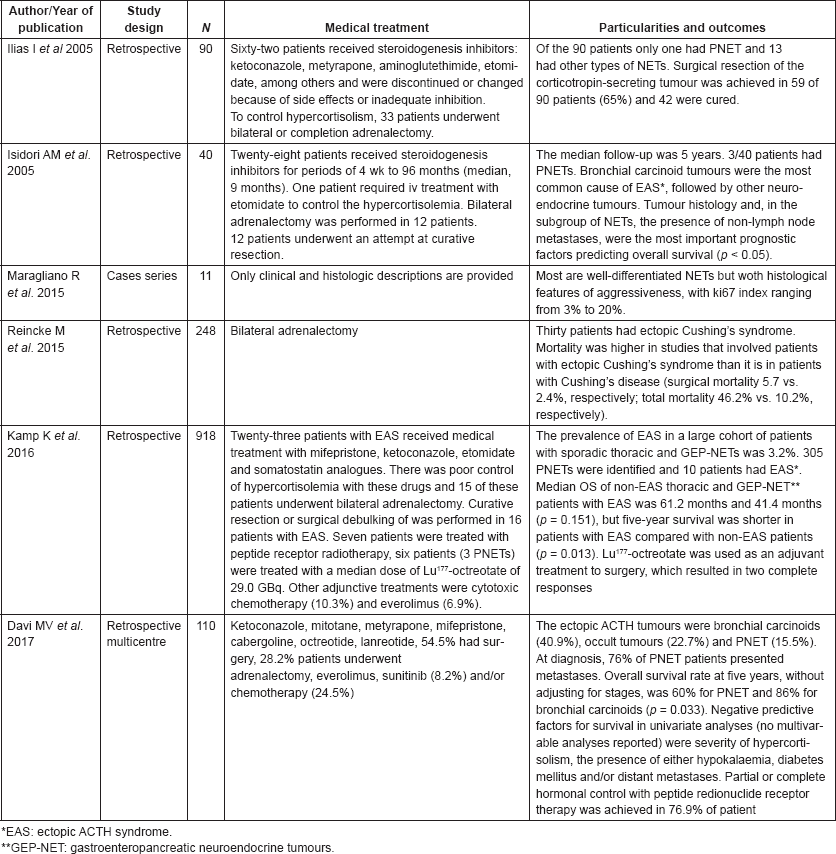
Table 2 shows studies that evaluated Cushing’s syndrome in patients with PNETs. The treatment aim for ectopic CS associated with PNETs was the maximum biochemical control of hypercortisolaemia, often combined with antitumour therapy. Medical treatment for CS mostly relies on steroidogenesis inhibitors (ketoconazole, mitotane and metyrapone). The CS control with these drugs is usually partial and transitory. In addition, the steroidogenesis inhibitors are associated with a high rate of hepatotoxicity. In our experience, bilateral adrenalectomy is often the only effective intervention to control CS from ectopic-ACTH neuroendocrine tumours. Likewise experts tend to indicate bilateral adrenalectomy to treat severe cases of CS not properly controlled with medical treatment [4]. Surgery debulking can be recommended with the aim of tumour for symptom control; hepatic embolization and radiofrequency ablation are also reasonable therapeutic options, although there are no data about their efficacy in controlling CS from ACTH-producing PNETs. In patients with advanced, surgically non-resectable or progressive PNET, systemic treatment can be performed [12]. Treatment of metastatic disease may reduce neoplastic lesions and control the ACTH and cortisol production [13]. However, there are few data about the effects of systemic therapies to treat CS associated with NET.
Somatostatin analogue, octreotide and lanreotide can be used to control neoplastic growth and ACTH ectopic production in tumours with somatostatin receptors expression, which is found in 80% of PNET [14]. Acting in somatostatin receptor subtype-2, the somatostatin analogue-controlled ACTH levels in a case report [15]. But given that ACTH-producing PNET tend to be aggressive, somatostatin analogues, in our experience, do not provide long-term tumour or symptom control. Targeted therapy represented by everolimus and sunitibe have been approved for the treatment of advanced well-differentiated PNETs. The RADIANT-3 trial showed that everolimus provided a 11-month median progression-free survival benefit (PFS) versus 4.6 months in the placebo group [16]. Another phase-III study demonstrated that sunitinib led to a median PFS of 11.4 months versus 5.5 months in the placebo group [17]. In terms of symptom and biochemical control, small case series showed the benefit of everolimus to control hypoglycaemia caused by metastatic insulinomas [18] and to improve carcinoid symptoms in patients with midgut tumors, [19] but data on the symptom control of ACTH-producing PNETs are lacking. Other treatment modalities include the use of systemic chemotherapy and interferon. Various single and combinatory cytotoxic chemotherapeutic agents have been used in the treatment of PNET, but there is little data on the best form of treatment for ACTH-producing PNET.
In our institution, which is funded by the Brazilian Unified Health System, targeted therapies and peptide receptor radionuclide therapy for NET are not available. Instead, systemic chemotherapy is the only reimbursed option for metastatic PNET. Bajetta et al conducted a phase-II trial of patients with metastatic neuroendocrine tumours, demonstrating a response rate of 30 among the well-differentiated tumours [20]. CApOx is the standard chemotherapy regimen for PNET in our institution and our retrospective data showed that the use of CApOX in patients with metastatic PNET offered a median disease progression of 9.8 months, partial objective response of 29% and stable disease in 71% of cases [21] . However, there are very limited data on the efficacy of chemotherapy to treat hormonal-syndromes associated with functioning PNETs. As demonstrated in our case, the CapOx, an inexpensive treatment, was able to control not only the tumour but also its ACTH production, as evidenced by the improvement in skin and nail hyperpigmentation. This had a positive impact on the patient’s quality of life in terms of improved self-esteem resulting from the normalisation of his skin colour.
Table 2. Cases series dealing with ectopic ACTH secretion with information on patient treatment and study outcomes.
Conclusion
Because of the rarity of ACTH-secreting PNETs, evidence-based treatment decisions are lacking, with the only source of scientific data being case series and reports. The experience of this case demonstrates that CApOX, an inexpensive regimen, may benefit patients with this tumour and represents a good option in settings of limited resources.
Conflicts of Interest
The authors declare no conflict of interest.
References
1. Ramage JK, Ahmed A, and Ardill J, et al (2012) Guidelines for the management of gastroenteropancreatic neuroendocrine (including carcinoid) tumors (NETs) Gut 61 6–32 https://doi.org/10.1136/gutjnl-2011-300831
2. Yao JC, Hassan M, and Phan A, et al (2008) One hundred years after “carcinoid”: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States J Clin Oncol 26 3063–72 https://doi.org/10.1200/JCO.2007.15.4377 PMID: 18565894
3. Hallet J, Law CH, and Cukier M, et al (2015) Exploring the rising incidence of neuroendocrine tumors: a population-based analysis of epidemiology, metastatic presentation, and outcomes Cancer 121 589 https://doi.org/10.1002/cncr.29099
4. Davi MV, Cosaro E, and Piacentini S, et al (2017) Prognostic factors in ectopic Cushing’s syndrome due to neuroendocrine tumors: a multicenter study Eur J Endocrinol 176 451–459 https://doi.org/10.1530/EJE-16-0809 PMID: 28183788
5. Ito T, Sasano H, and Tanaka M, et al (2010) Epidemiological study of gastroenteropancreatic neuroendocrine tumors in Japan J Gastroenterol 45 234–243 https://doi.org/10.1007/s00535-009-0194-8 PMID: 20058030
6. Tadokoro R, Sato S, and Otsuka F, et al (2016) Metastatic pancreatic neuroendocrine tumor that progressed to ectopic adrenocorticotropic hormone (ACTH) syndrome with growth hormone-releasing hormone (GHRH) production Intern Med 55(20) 2979–2983 https://doi.org/10.2169/internalmedicine.55.6827 PMID: 27746436 PMCID: 5109566
7. Maragliano R, Vanoli A, and Albarello L, et al (2015) ACTH-secreting pancreatic neoplasms associated with Cushing syndrome clinicopathologic study of 11 cases and review of the literature Am J Surg Pathol 39(3) 374–382 https://doi.org/10.1097/PAS.0000000000000340
8. Yao WQ, Wu X, and Li GD, et al (2015) ACTH-secreting pancreatic neuroendocrine carcinoma with ovarian and pelvic metastases causing Cushing’s syndrome: a case report Int J Clin Exp Pathol 8(11) 15396–15401
9. Wajchenberg BL, Mendonca BB, and Liberman B, et al (1994) Ectopic adrenocorticotropic hormone syndrome Endocr Rev 15 752–787 PMID: 7705280
10. Kondo T, Matsuyama R, and Ashihara H, et al (2010) A case of ectopic adrenocorticotropic hormone-producing pancreatic neuroendocrine tumor with multiple liver metastases Endo J 57(3) 229–236 https://doi.org/10.1507/endocrj.K09E-179
11. Wajchenberg BL, Mendonca BB, and Liberman B, et al Ectopic adrenocorticotropic hormone syndrome Endocr Rev 15 752–787 PMID: 7705280
12. Rajeev SP, McDougall S, and Terlizzo M, et al (2014) Evolution in functionality of a metastatic pancreatic neuroendocrine tumour (pNET) causing Cushing’s syndrome: treatment response with chemotherapy BMC Endocr Disord 14 70 https://doi.org/10.1186/1472-6823-14-70
13. Patel FB, Khagi S, and Dalin KP, et al (2013) Pancreatic neuroendocrine tumor with ectopic adrenocorticotropin production: a case report and review of literature Anticancer Res 33 4001–4006 PMID: 24023341
14. Clark ES and Carney JA (1984) Pancreatic islet cell tumor associated with Cushing’s syndrome Am J Surg Pathol 8(12) 917–924 https://doi.org/10.1097/00000478-198412000-00004 PMID: 6097131
15. Doi M, et al. (2003) Octreotide-Sensitive Ectopic ACTH Production by Islet Cell Carcinoma With Multiple Liver Metastases Endocr J 50 135–143 https://doi.org/10.1507/endocrj.50.135 PMID: 12803233
16. Yao JC, Shah MH, and Ito T, et al (2011) Everolimus for advanced pancreatic neuroendocrine Tumors N Engl J Med 364 514–523 https://doi.org/10.1056/NEJMoa1009290 PMID: 21306238 PMCID: 4208619
17. Raymond E, Dahan L, and Raoul JL, et al (2011) Sunitinib malate for the treatment of pancreatic neuroendocrine tumors N Engl J Med 364 501–513 https://doi.org/10.1056/NEJMoa1003825 PMID: 21306237
18. Kulke MH, Bergsland EK, and Yao JC, et al (2009) Glycemic control in patients with insolinoma treated with everolimus N Engl J Med 360 2 https://doi.org/10.1056/NEJMc0806740
19. Riechelmann RP, et al (2017) Guidelines for the management of neuroendocrine tumours by the Brazilian gastrointestinal tumour group ecancer 11 716 https://doi.org/10.3332/ecancer.2017.716
20. Bajetta E, Catena L, and Procopio G, et al (2007) Are capecitabine and oxilaplatine (XELOX) suitable treatments for progressing low-grade and high-grade neuroendocrine tumors? Cancer Chemother Pharmacol 59 637–642 https://doi.org/10.1007/s00280-006-0306-6
21. Ferrarotto R, Testa L, and Riechelmann RP, et al (2013) Combination of capecitabine and oxaliplatin is an effective treatment opcion for advanced neuroendocrine tumors Rare Tumors 5(3) e35 https://doi.org/10.4081/rt.2013.e35 PMCID: 3804810