CD19(➕) CD56 (–) myeloma arising in a patient who failed two courses of immunosupressive therapy for aplastic anaemia
Nigel P Murray1,2, M Amparo Ruiz3 and G Maximiliano Miranda1
1Service of Medicine, Hospital Carabineros of Chile, Nuñoa, 7770199 Santiago, Chile
2Faculty of Medicine, University Finis Terrae, Providencia, 7501015 Santiago, Chile
3Service of Pathology, Hospital Carabineros of Chile, Nuñoa, 7770199 Santiago, Chile
Correspondence to: Nigel P Murray. E-mail: nigelpetermurray@gmail.com
Abstract
Patients diagnosed with severe aplastic anaemia and without a compatible bone marrow transplant donor are treated with immunosuppressive therapy. These patients are found with time to develop a clonal disease such as myelodysplasia or paroxysmal nocturnal haemoglobinuria. However, the development of plasma cell dyscrasias is rare. We report the case here of a patient treated with immunosuppressive therapy who went on to develop myeloma 11 months after being diagnosed with severe aplastic anaemia. We include here a review of the literature.
Keywords: aplastic anemia, myeloma, immunosuppressive therapy
Copyright: © the authors; licensee ecancermedicalscience. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (
Published: 14/02/2017; Received: 18/10/2016
Introduction
In patients without a compatible sibling or unrelated donor standard immunosuppressive therapy (IT) with anti-thymocyte globulin (ATG) and cyclosporine A (CyA) produces excellent results with a reported response rate of 58–77% and a survival rate of 58% at 11 years [1–4]). The most serious complication of IT is the development of clonal disease, including the development of myelodysplasia and expansion of paroxysmal nocturnal haemoglobinuria (PNH) clones.
Immunophenotypic analysis with anti-CD19 and anti-CD56 can clearly distinguish between normal plasma cells and myeloma cells. Normal plasma cells are CD19 (➕) CD56 (-), whereas in myeloma approximately 65% are CD19 (-) CD56 (➕), 30% CD19 (-) CD56 (-) and 5% CD19 (➕) CD56 (➕) [5, 6]. We present a case of myeloma expressing the rare phenotype CD19 (➕) CD56 (-) in a 15-year-old female patient occurring six months after failing to respond to a second cycle of ATG-methylprednisone-cyclosporine for aplastic anaemia.
Clinical case
A 15-year-old girl presented with a three month history of extensive bruising and hypermenorrhea of up to 18 days. A full blood count showed a haemoglobin of 3.5 g/dL mean corpuscular volume of 111.6 fL, platelets of 6000/mm3, white cell count of 1500/mm3, an absolute neutrophil count of 450/mm3, and reticulocyte count of 0.5%. A provisional diagnosis of aplastic anaemia was made, and the supplementary testing showed; tests for hepatitis A, B, C, and HIV negative, antinuclear, anti-DNA antibodies negative, negative for Epstein-Barr virus and cytomegalovirus. Immunoglobulin (Ig) A 77 mg/dL (normal range 70–400), IgG 773 mg/dL (normal range 700–1600), and IgM 54 mg/dL (40–230). CD55/CD59 expression normal, bilirubin 0.37 (normal range <1.0), ferritin 31 (normal range 10–150), and lactate dehydrogenase 209 IU (normal range 100–250).
Bone marrow biopsy showed less than 20% cellularity. There were immature and precursor cells of the erythroid and myeloid series in central accumulations and paratrabecular location with very few mature forms. No megakaryocytes were observed.
Staining for myeloperoxidase showed positivity in 30% of immature cells which were arranged as paratrabecular sheets, while CD34 staining cells were less than 5%, with a central focus of immature cells staining positive for both CD34 and myeloperoxidase. Staining for PNCA was less than 1% and there was no evidence of fibrosis or lymphoid follicular formation (Figure 1). Bone marrow cytogenetics revealed a normal 46XX karyotype.
A diagnosis of aplastic anaemia was made. A suitable bone marrow donor was unavailable and hence immunosuppressive therapy with anti–thymocyte globulin, methylprednisone, and cyclosporine was started. There was no response at six months and a second cycle was repeated, again failing to achieve a response.
Eleven months after the initial presentation, the patient presented with high fever of 40ºC, and bilateral cervical adenopathy without any other localising signs. A CT scan of the neck showed bilateral cervical and jugular chain adenopathy of up to 22 mm with necrosis. Empirical antibiotic therapy was started with amikacin and the combination antibiotic piperacillin–tazobactam with platelet support (hydrocortisone and chlorpheniramine prophylaxis was administered with platelet transfusions). Blood and urine cultures were persistently negative. With the adenopathy increasing in size and to exclude a lymphoproliferative disease, an excisional biopsy of the node was performed along with a repeat bone marrow biopsy.
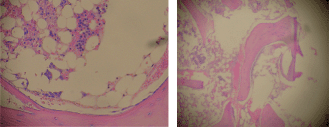
Figure 1. Bone marrow biopsy (H & E), 20% cellularity (magnification x 100).
The biopsy of the node showed a conserved structure but with loss of the lymphoid follicles. There was 70% of the lymph node parenchyma being replaced with an infiltration of morphologically mature plasma cells as well as loss of the normal architecture (Figure 2, 3).
The infiltrating cells were CD38 positive (Figure 4) and CD19 positive (Figure 5), with a kappa–lambda ratio of 1:4 (Figure 6–7)
The bone marrow biopsy (Figure 8) showed a variable cellularity occupying between 10–90% of the intertrabecular spaces with over 70% being plasma cells, diffusely infiltrated between a few myeloid precursors. Immunocytochemistry showed a lambda:kappa ratio of 9:1 (Figure 9, 10), the plasma cells being negative for CD20 and positive for CD19, CD38, and CD138, and 20% of plasma cells being positive for Bcl-2. Flow cytometry of the bone marrow showed that 23% of the nucleated cells expressed CD45, CD38i, CD138m, CD19, and cytoplasmic lambda light chain. The cells were CD56, CD117, CD20, CD22, and HLA–DR negative (Figure 11).
There was a hyper–gammopathy of 10 g/dL (normal range 3.0–4.5), with an IgA lambda monoclonal protein of 4.20 g/dL, total IgG 535m g/dL, and total IgM 32 mg/dL, both IgG and IgM being decreased, and a positive Bence-Jones protein. Serum creatine and calcium levels were normal and a skeletal survey showed a few small osteolytic lesions in the lateral skull x–ray.
A diagnosis of plasma cell dyscrasia consistent with myeloma was made. Because of the severe cytopaenia, dexamethasone 40 mg days 1–4 with a 10 day cycle was started. After six cycles, the Bence-Jones protein was undetectable, and the monoclonal protein had decreased to 2.30 g/dL. However, the patient presented with septic shock and died.

Figure 2. Low power biopsy ganglia (x 10).
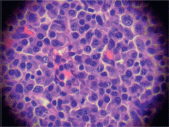
Figure 3. Infiltration by plasma cells (x 400).
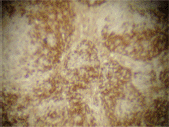
Figure 4. Plasma cells CD38 (➕) (x 10).
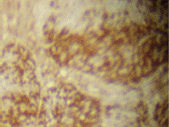
Figure 5. Plasma cells CD19 (➕) (x 10).
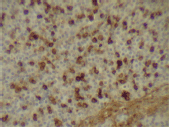
Figure 6. Kappa expression (x 400).
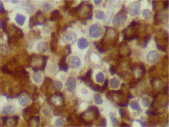
Figure 7. Lambda expression (x 600).
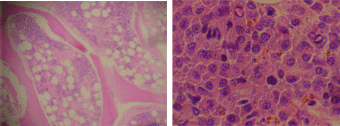
Figure 8. Bone marrow biopsy showing hypercellular features and infiltration by plasma cells (x 400).

Figure 9. Bone marrow biopsy stained for kappa (x 400).
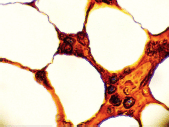
Figure 10. Bone marrow stained for lambda (x 1000).
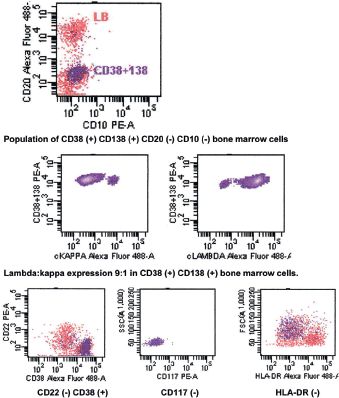
Figure 11. Flow cytometry of bone marrow showing CD56 (-) CD19 (➕) phenotype.
Discussion
There are only a few cases in the literature of aplastic anaemia progressing to myeloma. Manoharan et al reported a case in 1981 [7], and a second case of acquired aplastic anaemia evolving into myelodysplasia and finally myeloma was reported in 1991 [8].
There are cases reported of aplastic anaemia associated with a minimal serum M–protein [9] or a marked reactive plasmacytosis secondary to a drug reaction [10], autoimmune disease [11], or infection [12].
In the case we describe here, we consider from the initial findings that myeloma was not present. Initial bone marrow biopsy revealed no infiltration with plasma cells (with we recognising the fact that myeloma can also present with severe marrow hypoplasia) [13]. There was no serum paraprotein, and the patient had normal immunoglobulin levels. There was no evidence of a hypoplastic myelodysplasia at the initial biopsy as suggested by the low PNCA and CD34 expression [14]. The clinical and pathological findings were consistent with acquired aplastic anaemia.
After IT failure, the patient was considered to have developed myeloma based on the 2015 NCCN guidelines. Sahara et al [15] first reported a case of CD19 (➕) CD56 (–) myeloma. The CD38, CD138 phenotype combined with low levels of CD45 expression, excluded the possible contamination with CD19 (➕) CD56 (–) lymphoid cells. The cellular morphology, monoclonal plasma cell proliferation, monoclonal gammopathy, Bence-Jones protein, and osteolytic lesions supported the diagnosis of myeloma. Although the cervical lymph nodes were infiltrated by plasma cells with the same immunophenotype as the bone marrow, this was considered to be extra–medullar extension rather than a plasmacytoma.
Studies have reported that low CD56 expression is associated with extensive bone marrow and blood involvement; a more aggressive disease and a significantly shorter survival [16]. It has been recommended that CD56 (–) myeloma be treated intensively to overcome this poor response [17]. However, treatment options were limited because of the severely hypocellular bone marrow, dexamethasone produced an initial response with decreases in the M-protein and disappearance of the Bence-Jones protein. Although showing a response, the patient died as a result of septic shock and acute multiorgan failure.
The differential diagnosis of plasmablastic lymphoma was also considered. However, the lesion being lytic with bone marrow involvement, having morphological characteristics of myeloma rather than a large cell lymphoma, having a monoclonal paraprotein with a positive Bence-Jones protein, and finally being HIV negative, all supported the diagnosis of myeloma rather than lymphoma. Plasmablastic lymphoma is an uncommon diffuse large B–cell lymphoma with a predilection for individuals HIV positive or with immunosuppression. Biopsy of the lymph node in plasmablastic lymphoma shows poorly differentiated plasma cells with low or absent B–cell markers (CD19, CD20, and CD22, and surface/cytoplasmic immunoglobulin), and positivity for plasma cell markers such as CD38 and CD138 [16].
Conclusion
In summary, our case presents the rare CD19 (➕) CD56 (–) phenotype of myeloma arising in a patient treated by IT for aplastic anaemia. Treatment of what is considered to be an aggressive form of myeloma is severely limited in patients with poor marrow reserve and lack of a transplant donor. With only a few cases reported of myeloma arising from aplastic anaemia treated with IT reported in the literature, it is difficult to confirm this association. However, in this case report there did not appear to be evidence of myeloma pre-IT whereas 18 months after the initial diagnosis and two cycles of IT, the evidence suggested the rare phenotype of myeloma.
Ethical considerations
This case study was approved by the Hospital de Carabineros Ethical Committee, complying with the Chilean Law 20,458 of patients’ rights.
Conflicts of interest
The authors report no conflicts of interest.
Acknowlegdments
The authors would like to thank Mrs Ana Maria Palazuelos for her help in the writing of this manuscript.
References
1. Bacigalupo A et al (2000) Antilymphocyte globulin, cyclosporin, prednisone and granulocyte stimulating factor for severe aplastic anemia: an update of the GITMO/EBMT study on 100 patients. European Group for Blood and Marrow Transplantation (EBMT) Working Party on Svere Aplastic Anemia and the GruppoItalianoTrapianti di Midolio Osseo (GITMO) Blood 95(6) 1931–4 PMID: 10706857
2. Frickhofen N et al (2003) Antothymocyte globulin with or without cyclosporin A: 11 year follow up of a randomized trial comparing treatments of aplastic anemia Blood 101(4) 1236–42 DOI: 10.1182/blood-2002-04-1134
3. Rosenfeld S et al (2003) Antithymocyte globulin and cyclosporin for severe aplastic anemia: associaiton between hematologic response and long term outcome JAMA 289(9) 1130–5 DOI: 10.1001/jama.289.9.1130 PMID: 12622583
4. Tichelli A et al (2011) A randomized controlled study in patients with newly diagnosed severe aplastic anemia receiving antithymocyte globulin (ATG), cyclosporine, with or without G–CSF: a study of the SAA Working Party of the European Group for Blood and Marrow Transplantation Blood 117(17) 4434–41 DOI: 10.1182/blood-2010-08-304071 PMID: 21233311
5. Harada H et al (1993) Phenotypic difference of normal plasma cells from mature mieloma cells Blood 81(10) 2658–63
6. Rawstrom AC et al (1997) Circulating plasma cells in multiple mieloma: characterization and correlation with disease stage Br J Haematol 97(1) 46–55
7. Manoharan A, Horsley R and Pitney WR (1981) Myelomatosis in aplastic anemia–a true association or fortuitous occurence? Pathology 13(4) 771–3 DOI: 10.3109/00313028109086651 PMID: 7335384
8. Herraez Garcia J et al (1991) Evolution of acquired aplastic anemia to myelodysplasia and posterior multiple myeloma An Med Interna 8(6) 313
9. Yokoh K et al (1990) Aplastic anemia associated with minimal serum M–protein treated successfully with repeated bolus methlyprednisona therapy Rinsho Ketsueki 31(4) 506–10 PMID: 2381070
10. Yamamoto M et al (2012) Marked reactive plasmacytosis accompanied by drug eruption in a patient with aplastic anemia Rinsho Ketsueki 53(5) 526–30 PMID: 22728555
11. Hanai J et al (1980) Aplastic anemia with hypergammaglobulina, systemic plasmacytosis and postive RA trst–histopathological study on an autopsy case Nihon Ketsueki Gakkai Zasshi 43(4) 689–99 PMID: 7223335
12. Nishimoto Y et al (1987) Hepatitis associated aplastic anemia with systemic plasmacytosis Acta Pathol Jpn 37(1) 156–66
13. Medhi K et al (2008) Multiple mieloma presenting with co–existing severe marrow hupoplasia. Indian J PatholMicrobiol 51(4) 543–5 PMID: 19008591
14. Orazi A et al (1997) Hypoplastic myelodysplastic síndromes can be distinguished from acquired aplastic anemia by CD34 and PCNA immunostaining of bone marrow biopsy specimens Am J Clin Pathol 107(3) 268–74 DOI: 10.1093/ajcp/107.3.268 PMID: 9052376
15. Sahara N et al (2000) Multiple myeloma expressing CD19 ( ) CD56 (–) phenotype Am J Hematol 64(4) 311–13 PMID: 10911386
16. Sahara N and Takeshita A (2004) Prognostic significance of surface markers expressed in multiple myeloma: CD56 and other antigens Leuk Lymphoma 45(1) 61–5 DOI: 10.1080/1042819031000149377 PMID: 15061198
17. Lin P, Owens R and Tricot G(2004) Flow cytometric immunophenotype analysis of 306 cases of multiple mieloma Am J Clin Pathol 121(4) 482–8 DOI: 10.1309/74R4TB90BUWH27JX PMID: 15080299
18. Kravetz JD et al (2006) Plasmablastic lymphoma presenting as an arm mass in an individual negative for HIV: a case report Clin Lymphoma Myeloma 6(6) 493–5 DOI: 10.3816/CLM.2006.n.032 PMID: 16796782





