Repurposing Drugs in Oncology (ReDO)—Propranolol as an anti-cancer agent
Pan Pantziarka1,2, Gauthier Bouche1, Vidula Sukhatme3, Lydie Meheus1, Ilse Rooman1,4 and Vikas P Sukhatme3,5
1Anticancer Fund, Brussels, 1853 Strombeek-Bever, Belgium
2The George Pantziarka TP53 Trust, London, UK
3GlobalCures, Inc, Newton MA 02459, USA
4Oncology Research Centre, Vrije Universiteit Brussel, 1090 Brussels, Belgium
5Beth Israel Deaconess Medical Center and Harvard Medical School, Boston, MA 02215, USA
Correspondence to: Pan Pantziarka. Email: pan.pantziarka@anticancerfund.org
Abstract
Propranolol (PRO) is a well-known and widely used non-selective beta-adrenergic receptor antagonist (beta-blocker), with a range of actions which are of interest in an oncological context. PRO displays effects on cellular proliferation and invasion, on the immune system, on the angiogenic cascade, and on tumour cell sensitivity to existing treatments. Both pre-clinical and clinical evidence of these effects, in multiple cancer types, is assessed and summarised and relevant mechanisms of action outlined. In particular there is evidence that PRO is effective at multiple points in the metastatic cascade, particularly in the context of the post-surgical wound response. Based on this evidence the case is made for further clinical investigation of the anticancer effects of PRO, particularly in combination with other agents. A number of trials are on-going, in different treatment settings for various cancers.
Keywords: Propranolol, beta-blockers, drug repurposing, ReDO project
Copyright: © the authors; licensee ecancermedicalscience. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Published: 12/10/2016; Received: 06/06/2016
Introduction
Propranolol (PRO) is a commonly used non-selective beta-adrenergic receptor antagonist used in the treatment of hypertension, angina, anxiety, cardiac arrhythmia, hyperthyroidism, essential tremor and as a prophylaxis against migraine, variceal bleeding and myocardial infarction. First developed in the 1960s, the drug is now available globally in generic form and is on the WHO List of Essential Medicines. The drug is available in both standard and extended-release tablet formulations, as an oral solution and also as intravenous injection. Common trade names include Inderal, Angilol, Syprol, Ciplar. A special oral formulation, (Hemangiol in Europe, Hemangeol in the USA), has also been licensed by the EMA and FDA, for the treatment of infantile hemangioma. There is also clinical trial data supporting the off-label use of PRO in a number of conditions, including haemorrhage, sepsis and hypermetabolic syndrome associated with severe burns [1], akathisia associated with Alzheimer’s disease or psychosis [2], and aggression associated with brain injury or disease [3].
In addition to clinical use in these varied indications there is accumulating evidence that propranolol has potent anti-cancer effects, as evidenced by in vitro, in vivo and a range of clinical data.
Current Usage
Dosage
The PRO dose varies by indication. The anti-hypertensive dose is in the range 160 – 320 mg/day, starting at 80 mg and increasing as required to a maintenance dose that is generally 160 mg – 240 mg, in divided doses or as once a day use of extended release tablets. For angina the dose is 120 – 240 mg/day. Migraine prophylaxis is in the range 80 – 240 mg/day [4]. The dose in infantile hemangioma is 1 mg/kg/day for 1 week, then 2 mg/kg/day for 1 week and then 3 mg/kg/day as a maintenance dose for 6 months.
Toxicity
Common side effects include insomnia, fatigue, cold extremities and Raynaud’s syndrome. Less common side effects include nausea, vomiting, diarrhoea. Rarely PRO is associated with heart failure, heart block, hypotension, worsening of symptoms in psoriasis, asthma and psychosis. In general the initiation of PRO may lead to initial, mild adverse effects which resolve during dose titration to a maintenance dose. Sudden termination of treatment is not advised, particularly for patients suffering ischaemic heart disease – in such cases the dose should be tapered rather than stopped abruptly. In general however, PRO has a good toxicology profile and can be used for long-term treatments of many years duration [5].
PRO is contra-indicated in patients suffering from hypotension, asthma, uncontrolled heart failure, severe peripheral artery disease, metabolic acidosis and cardiogenic shock. It is not recommended in pregnancy and during lactation.
Pharmacokinetics
PRO is highly lipophilic and undergoes rapid absorption in the gastrointestinal tract and more than 90% undergoes plasma protein absorption. Excretion is primarily renal, though 1 – 4% of an oral or IV dose of the drug appears in faeces as unchanged drug and metabolites [6]. There is wide distribution to tissues, particularly lungs, liver, kidneys, and heart. Bioavailability after oral dosing is in the range 25 – 35% due to extensive first-pass hepatic clearance, although there is considerable inter-patient variability [7]. Bioavailability can be increased by concomitant food intake, with a mean increase of around 50% reported when taken after a protein-containing meal, however other parameters (time to maximum concentration, half-life etc.) are unchanged [8].
Peak plasma concentrations occur 1.5 – 3 hours following oral dosing, with a plasma half-life of around 4 hours following single dose or around 10 hours for extended release tablets. Mean peak plasma concentration following a single oral dose of 40 mg, in fasting conditions, is 38 ng/ml (0.12 μM) [9]. A single oral dose of 160 mg produced a peak in the range 200 – 245 ng/ml (0.77 – 0.96 μM), while the extended release tablets produced a peak in the range 18 – 50 ng/ml (0.07 – 0.19 μM) [10]. PRO crosses the blood brain barrier [11]. There is some evidence that the effects of PRO are dependent on the plasma concentrations that are achieved, including in the treatment of rare benign tumours in children [12].
Hepatic metabolism of PRO involves multiple pathways in the cytochrome P450 system (CYP2D6, 1A2 and 2C19), and therefore a range of drug interactions are possible [4, 6]. For example, concurrent cimetidine increased the area under the curve (AUC) and doubled peak plasma levels of PRO [13].
Caution is advised when PRO is used with calcium-channel blocking drugs, particularly IV verapamil, in patients with severe cardiomyopathy, congestive heart failure, or recent myocardial infarction due to the negative inotropic and chronotropic action of these drugs [14].
Pre-clinical Evidence in Cancer - In Vitro and In Vivo
Investigation of the possible anticancer properties of PRO began in the late 1970s, primarily with regards to elucidating the roles of catecholamines in carcinogenesis and in identifying beta-adrenergic receptor binding sites in different tissues [15–17].
Leukaemia
One of the first findings that PRO may be of some benefit in cancer treatment was reported by Ramu et al, who, in 1984, published a report that looked at the activity of a range of drugs in reversing in vitro drug resistance in the P388/ADR murine leukaemia cell line [18]. PRO was shown to have moderate effects in restoring sensitivity to doxorubicin in P388/ADR cells, but showed no evidence of additional effects in the parental P388 cell line. Similarly, Tsuruo et al showed that 10 μM of PRO significantly (P < 0.05) enhanced the cytotoxicity of vincristine and doxorubicin in resistant P388/VCR and P388/ADR cell lines respectively [19]. Other investigators have also reported similar effects in reversing resistance in multi-drug resistant human epidermoid KB carcinoma cell lines [20].
Hajighasemia and Mirshafiey investigated the cytotoxicity of PRO against Molt-4 and Jurkat human leukaemia and the U937 monocyte cell lines [21]. They showed that the viability of all three cell lines was dose- and time-dependently reduced by PRO above concentrations of 200 μM.
An investigation by Lamkin et al in a murine model of acute lymphoblastic leukaemia (ALL) found that chronic stress enhanced tumour growth and dissemination and that the effect could be inhibited by PRO [22]. PRO has also been shown to inhibit the expression of the tissue remodelling factor matrix metalloproteinase-2 (MMP-2) and the pro-angiogenic vascular endothelial growth factor (VEGF) in human leukaemia cell lines [23].
Breast
Shakhar and Ben-Eliyahu reported on the influence of beta adrenergic agonists and antagonists on natural killer (NK) cell number and activity in F344 rats inoculated with highly metastatic MADB106 syngeneic mammary adenocarcinoma cells [24]. Injection of the beta-adrenergic agonist metaproterenol (MP) led to a transient increase in NK cell numbers which returned to baseline within one hour; however, there was a concomitant decrease in NK activity over the same period. The beta-adrenergic antagonists nadolol and PRO were able to suppress these effects. Furthermore, treatment with MP was associated with a 10-fold increase in the number of injected tumour cells retained in the lungs 1-day post-inoculation, and a corresponding increase in pulmonary metastatic lesions three-weeks post-inoculation. These effects were dose-dependent and reversible by co-treatment with the non-selective beta-blocker nadolol.
Benish et al investigated the effect of inhibiting cyclo-oxygenase-2 (COX-2) and of blocking beta-adrenergic receptors on post-surgical immune function and metastatic tumour growth [25]. F344 rats underwent laparotomy and were injected with syngeneic MADB106 cells. Rats pre-treated with COX-inhibitors (SC560, indomethacin, etodolac, or celecoxib) or vehicle were compared to non-surgically treated controls to assess the impact on tumour cell retention in the lungs (LTR), and it was shown that all surgically treated animals had an elevated rate of LTR compared to non-surgically treated, but that COX-2 inhibition (indomethacin, etodolac, or celecoxib, administered one hour prior to surgical incision) significantly attenuated the increase compared to the vehicle treated group. Other experiments combined etodolac and PRO, (both administered one hour prior to incision), and again showed that LTR was reduced by either treatment alone and in combination, and that chronic and acute treatment had similar outcomes. Finally, it was shown that surgery was also associated with reduced NK cell cytotoxicity, which could also be reversed by the combination of etodolac and PRO. Subsequent work by the same group reproduced similar results and additionally showed that combination treatment with the immunostimulant CpG-C increased the effect of the PRO etodolac combination [26].
The effect of sympathetic nervous system signalling was investigating in a murine breast cancer model by Sloan et al [27]. BALB/c mice were subjected to two hours per day of restraint, shown previously to induce catecholamine-mediated stress, or control conditions for five days prior to injection of syngeneic 66cl4 mammary carcinoma cells. Stressed animals showed reduced weight and a 38-fold increase in the rate of metastasis (P = 0.04), both in terms of increased number and size of metastases compared to unstressed controls. Increased stress was not associated with significant changes in primary tumour growth. PRO treatment had no effect on metastatic growth in control animals but completely inhibited the enhanced increase in metastases in stressed animals (P < 0.001). Others have also investigated the role of norepinephrine in the metastatic process and reported a similar anti-metastatic activity of PRO in murine models of breast cancer [28–29].
Having first ascertained that low concentrations of PRO and 5-FU or paclitaxel increased the anti-proliferative and anti-angiogenic effects of these standard chemotherapeutic drugs in a panel of cancer and non-cancer cell lines. Pasquier et al studied the in vivo effects of the combination in NMRI nude mice orthotopically injected with MDA-MB-231 human triple negative breast cancer cells [30]. PRO, at a dose of 10 mg/kg, with paclitaxel increased median survival by 79% compared to paclitaxel alone (P = 0.0005) and PRO with 5-FU increased median survival by 19% (P = 0.0005).
The role of beta-adrenergic signalling in breast cancer metastasis to the bone was investigated by Campbell et al [31]. In vitro experiments showed that beta-adrenergic signalling upregulated RANKL expression in osteoblasts and that this increased MDA-MB-231VU human mammary carcinoma cell migration. An in vivo model, using the same cell line in athymic mice, showed that chronic stress or exogenous beta-adrenergic agonist isoproterenol increased both the number and area of osteolytic lesions compared to controls and that PRO, supplied ad libitum (0.5 g/L) via drinking water, could reverse this increase (P < 0.05).
In HER2-amplified breast cancer, Liu et al investigated the role of catecholamines and PRO on resistance to trastuzumab [32]. After showing a strong association between trastuzumab-resistance and beta2 adrenergic signalling in patient tissue samples, they showed that the catecholamines epinephrine and isoproterenol antagonised the anti-proliferative effect of trastuzumab both in vitro and in vivo. Additionally they showed that PRO could inhibit this antagonist effect, and resensitise resistant cells, both in vitro and in a xenograft model (PRO dosed at 2 mg/kg).
Melanoma
The impact of psychosocial stress on cancer growth in two murine model of cancers was studied by Hasegawa and Saiki [33]. Groups of mice were housed in different conditions to simulate the effects of social crowding on growth of syngeneic tumours (B16 melanoma in C57BL/6 mice and Meth A fibrosarcoma in BALB/c mice). Three housing conditions were used – isolated, grouped and over-crowded – and the effect on tumour growth assessed. B16 melanoma growth rates were increased most in the order over-crowded, isolated and grouped animals. Additionally another over-crowded cohort was administered PRO, at a dose of 30 ppm from days -21 to 21 after tumour implantation. This cohort showed reduced tumour growth dynamics, with the initial growth rate lower than the grouped (non-stressed) cohort and was significantly lower than either of the stressed groups. Organ weight was also assessed and a negative correlation detected between thymic mass and tumour mass (P < 0.05). Repeated experiments compared the effects of over-crowding versus isolation in additional cohorts of melanoma-bearing C57BL/6 mice and in sarcoma-bearing BALB/c mice. Over-crowding was shown to be more strongly associated with increased tumour growth and thymic atrophy than isolation.
Dal Monte et al investigated the role of beta3 adrenergic receptors in melanoma growth and vascularisation [34]. In some of the in vitro experiments PRO was used alongside two selective beta3 adrenergic receptor antagonists, SR59230A and L-748,337. PRO, at a concentration of 10 μM, with and without exogenous norepinephrine, significantly reduced proliferation (P < 0.001) and increased apoptosis (P < 0.01) of B16F10 cells compared to controls. Wrobel and Le Gal also showed that PRO had significant effects on proliferation and apoptosis in vitro on a panel of melanoma cell lines at a high concentration of 100 μM [35]. In vivo experiments using both primary and metastatic human melanoma tumours transplanted into immunodeficient (Nod SCID Gamma) mice showed that PRO at an average dose of 1.7 mg/day, tumour volumes were significantly lower (P < 0.001) than in untreated controls.
Glasner et al investigated the effects of surgical intervention on survival in two syngeneic mouse models, and on the impact of pre-operative PRO and etodolac, a COX-inhibitor, on survival [36]. B16 melanoma-bearing C57BL/6J mice were administered PRO, etodolac, PRO etodolac, or vehicle 30-minutes prior to amputation and/or laparotomy. Treatment with either drug singly showed no statistically significant difference with vehicle in terms of survival for any of the surgical options, whereas combined PRO etodolac significantly improved survival rates (P = 0.0345). In a Lewis Lung carcinoma model mice were pre-treated with IL-12 or vehicle 24-hours prior to amputation, treatment groups were further subdivided and treated with PRO etodolac or vehicle prior to surgery. All treatment groups showed significant increases in survival rates, although there were no differences in effect sizes between IL-12, IL-12 PRO etodolac and PRO etodolac treatments.
Ovarian
Lutgendorf et al noted that ovarian cancer patients with greater levels of social isolation and distress had greater levels of serum VEGF, associated with increased angiogenesis, in contrast to patients with lower levels of social isolation [37]. Subsequent in vitro investigation using the SKOV3 and EG ovarian carcinoma cell lines showed that norepinephrine, epinephrine, isoproterenol (a nonspecific beta-adrenergic agonist), and cortisol enhanced the production of VEGF in both cell lines. Pre-treatment with PRO, at a concentration of 1 μM, abolished this increase in VEGF [38]. Later work showed that in a murine SKOV3 model PRO was able to reduce isoproterenol-induced tumour growth [39].
Subsequent investigation showed that surgical stress, from a wound distant from the implanted tumour, was associated with increased primary tumour growth rate and the multiplicity of metastases in a two murine ovarian cancer models (HeyA8 and SKOV3ip1) but not in beta-adrenergic receptor-negative RMG-II mice [40]. Treatment with PRO via micro-osmotic pump starting seven days prior to surgical intervention inhibited the post-surgical increase in tumour growth rate and reduced the number of metastatic nodules.
Angiosarcoma
In light of the positive clinical experience of PRO in the treatment of infantile hemangioma, most recently confirmed in a large multi-centre randomised controlled trial [41], and evidence of beta-adrenergic receptor expression in a range of vascular tumours [42], a number of investigators have explored the potential benefit of PRO. Stiles et al used in vitro and in vivo models of hemangioendothelioma and angiosarcoma to investigate the impact of beta-blockade with PRO on cell proliferation, migration and apoptosis [43]. Using a panel of canine angiosarcoma, murine angiosarcoma and murine hemangioendothelioma cell lines, it was shown that 25 μM of PRO inhibited proliferation in all lines compared to untreated controls (P < 0.05). At a higher concentration of 100 μM, PRO induced apoptosis in all cell lines (P < 0.05), and showed synergistic action in cells treated with chemotherapy (cisplatin, busulfan, vincristine, or H2O2) with the exception of the murine hemangioendothelioma cell line. Finally, in a murine model of angiosarcoma PRO treatment at a dose of 20 mg/kg every 2 days led to a significant reduction of tumour growth compared to controls (tumour weight of 357 /-58 mg; N = 17 vs sham 984 /-92 mg; N = 15, P < 0.0001). Despite the reduction in tumour size, tumour sections from both sham and PRO conditions revealed active cell division, suggesting the need to employ combinatorial therapy with PRO.
Pasquier at al showed that immortalised and NRAS-transformed endothelial cells were sensitive to the anti-proliferative effects of PRO [44]. Furthermore, PRO was shown to have antagonistic or additive effects when combined with doxorubicin or paclitaxel, commonly used to treat angiosarcoma, but that the effect was synergistic in combination with vinblastine. In a 3D in vitro model BMST-Ras cells were allowed to form spheroids of ~600 μm in diameter before treatment was initiated. When used alone, 10 μM propranolol and 1 nM vinblastine significantly slowed down the growth of tumour spheroids, resulting in a 19 – 20% decrease in volume after 5 days of treatment as compared to untreated spheroids (P < 0.001). The combination of propranolol and vinblastine completely suppressed the growth of tumour spheroids, leading to a 59% decrease in volume after 5 days as compared to control spheroids (P < 0.001).
Neuroblastoma
Pasquier et al tested a panel of seven beta-adrenergic antagonists, alone and in combination with vincristine, on BE(2)-C and SHEP neuroblastoma cell lines in vitro [45]. Of the seven drugs tested the most potent anti-proliferative and anti-angiogenic agents were carvedilol, nebivolol and PRO. While these three agents did not significantly impact on the anti-cancer effects of a range of chemotherapeutics, they showed significant (P < 0.001) synergy with the vinca alkaloid vincristine. Similar results were obtained with vinblastine and paclitaxel, suggesting the effect was related to microtubule disruption. In vivo results, using a TH-MYCN transgenic mouse model of neuroblastoma, showed that PRO, at a dose of 50 mg/kg i.p., increased the rate of tumour regression induced by vincristine treatment (P < 0.05). Finally, analysis of survival times showed that mice treated with PRO and vincristine had a fourfold increase in median survival compared to treatment with vincristine alone (P < 0.0001). Carvedilol with vincristine seemed the most effective combination and resulted in sustained complete remission in one animal (of ten), which remained tumour-free until study completion (day 60).
A xenograft model of paediatric neuroblastoma, based on the BE(2)-C cell line, was used by Xu et al [46]. Animals with established tumours were treated with different doses of PRO – 2, 5 or 10 mg/kg – for nine days and compared to untreated controls. Animals were sacrificed on day 9 and tumour weights in the 2 mg/kg and 5 mg/kg groups were 36.6% (P < 0.002) and 34.4% (P < 0.005) lower than the control group. Tumour weights in the 10 mg/kg were 18.3% lower than control, a figure not statistically significant. Additional analyses showed that microvessel density (MVD) was lower in the treated groups than in controls (P < 0.01) and VEGF, MMP-2, and MMP-9 protein levels were significantly lower 5 mg/kg and 10 mg/kg groups (P < 0.05).
Wolter et al tested PRO against a panel of 15 neuroblastoma cell lines representing a range of genetic profiles [47]. PRO inhibited cell growth, reducing proliferation and viability, in all lines at IC50 values in the range 114 μM to 218 μM. It was also shown to be synergistic with SN-38, the active metabolite of irinotecan, with significantly reduced viability compared to treatment with either agent alone (P = 0.008 for PRO alone, P = 0.0009 for SN-38 alone). Using SK-N-AS cells in xenograft models, treatment with PRO at 2 mg/kg/day injected subcutaneously produced lower tumour volume at day 14 compared to controls, (P = 0.0246) and was associated with longer median survival (P = 0.0135).
Prostate
Work by Masur et al showed that norepinephrine, at a concentration of 10 μM, increased the migratory activity of PC3 human prostate carcinoma cells in vitro but had no influence on proliferation levels, and that PRO at the same dose significantly inhibited this increase [48]. Furthermore, athymic BALB/c mice injected with PC3 cancer cells were treated with norepinephrine, PRO or a combination, administered via micro-osmotic pumps, and the rate of primary and metastatic tumour growth was assessed. While neither treatment had significant effect on primary tumour growth, norepinephrine was associated with a significant increase (P = 0.014) in metastatic tumour growth compared to controls, and PRO treatment reduced metastatic tumour growth below controls (P = 0.009).
Brohée et al showed that in vitro PRO concentrations of 100 μM potentiated the anti-proliferative effect of rapamycin on human prostate cancer PC3 cells [49].
Pancreatic
Guo et al showed that PRO inhibited MMP-2, MMP-9 and VEGF in pancreatic cancer cell lines [50]. Zhang et al also reported that PRO was able to induce apoptosis in the PC-2 human pancreatic cancer cell line at concentrations of 100 μM [51], and reduce invasiveness in MIA PaCa-2 and BxPC-3 cell lines at the same concentration [52].
Kim-Fuchs et al used Panc-1 human pancreatic cancer cells to establish orthotopic tumours in BALB/c-Foxn1nu nude athymic mice [53]. Mice were subject to restraint or home cell conditions to mimic stress or control respectively. Stress, verified by changes in body weight and tissue catecholamine levels, was associated with an increased rate of pancreatic tumour growth by 11% ± 3 per day compared to control mice (P < 0.01) and increased tumour mass five-fold (7.5 mg ± 5 vs. 41 mg ± 13, P = 0.03). Additionally chronic stress was associated with metastatic spread in 50% of mice, compared to none in the controls. PRO treatment, via osmotic pump delivering 10 mg/kg/day, blocked the effect of stress on primary tumour growth (41 mg ± 13 vs. 21 mg ± 5), although there was no change in the metastatic rate in the time-frame of the experiment.
Partecke et al also investigated the impact of chronic stress on pancreatic cancer growth using C57BL/6 mice bearing orthotopic syngeneic 6606PDA tumours [54]. Stress was shown to be associated with immunosuppression and an increase in tumour angiogenesis and cancer cell invasion. Median survival in stressed mice was significantly reduced compared to unstressed control mice (52 days versus 66 days, P < 0.0058). Treatment with PRO, (given orally via drinking water using a concentration of 0.5 g/l of drinking water aiming at 30 – 60 mg/kg mouse/day), reduced tumour growth rates and increased survival compared to untreated controls (59 days versus 52 days in controls, P < 0.0058).
Colorectal
Masur et al investigated the effect of norepinephrine and PRO on the in vitro migratory activity of SW 480 colon carcinoma cells [55]. Where norepinephrine caused a dose dependent increase in the number of migrating cells above basal levels, treatment with PRO at the same concentrations (1 μM, 10 μM and 100 μM) abolished this increase. In contrast treatment with the selective beta1 adrenoreceptor antagonist atenolol did not interfere with the norepinephrine-induced increase in migratory cell numbers. Similarly, Coelho et al explored the impact of a number of beta adrenergic receptor agonists and antagonists on the proliferation of HT-29 colon adenocarcinoma cells [56] and reported the IC50 of PRO as 65.4 μM.
Chin et al also investigated the effect of selective beta2-adrenergic receptor antagonists, including PRO, in a panel of colorectal cancer cells lines [57]. They showed that PRO significantly inhibited the viability of SW620, Colo205, and HT29 cells (IC50 119.5, 86.38, and 69.1 μM, respectively). PRO induced G1-phase arrest and apoptosis in affected cell lines.
Lin et al showed that chronic restraint stress promoted tumour growth in xenograft models of colorectal cancer, and that this effect could be blocked, in vivo, using PRO at a dose of 2 mg/kg [58].
Head and Neck
Yang et al investigated the effects of norepinephrine on nasopharyngeal carcinoma cell lines (HONE-1, HNE-1, and CNE-1) [59]. Treatment of HONE-1 cells with norepinephrine upregulated levels of metalloproteinases (specifically MMP-2 and MMP-9) and VEGF and increased cell invasiveness. Treatment with PRO inhibited this increase in MMP-2, MMP-9 and VEGF. Inhibition by PRO of MMP-9 secretion has also been confirmed in human brain microvascular endothelial cells [60].
Bernabé and colleagues assessed the influence of norepinephrine and cortisol on oral squamous cell carcinoma cell lines (OSCC) [61]. They showed that norepinephrine and cortisol, at physiologically relevant levels, induced IL-6 production in SCC9, SCC15, and SCC25 cells and similar observations were made for isoproterenol in SCC9 and SCC25 cells. These effects were associated with an increase in cell proliferation and PRO, at a concentration of 1 μM, blocked this increase. Wolter et al also showed that PRO reduced the viability of SCC9, SCC17a, SCC25, and FaDu cell lines, and that it synergised with cisplatin and radiotherapy in treating SCC17a cells [62].
Other
Grzanna and co-workers showed, in vivo, that administration of PRO delayed tumour growth of LPC-1 plasmacytoma tumours in female BALB/c mice [63]. Mice were treated with doses of 0.6. 6 and 60 mg/kg/day, administered via osmotic pumps, and compared with untreated mice following subcutaneous injection of LPC-1 cells. All treated mice showed delayed tumour growth compared to untreated controls, in a dose-dependent manner. There was also a dose-dependent decrease in IgG plasma values, confirming the effect seen on tumour size. However, while treatment increased tumour growth latency, there was no difference in growth rates once tumours were established.
The effect of PRO on tumour perfusion was investigated by Bomber et al in a small 1986 study focused on enhancing uptake of Ga-67 to improve imaging of small tumours [64]. PRO was administered, at a dose of 10 mg/kg, 10 minutes prior to administration of radiolabelled gallium citrate. Analysis showed that PRO doubled tumour perfusion and decreased Ga-67 uptake in non-tumour tissues, thereby increasing the relative uptake in the tumour compartment. Subsequently Burton and Gray reported that a combination of norepinephrine and PRO enhanced the relative blood supply to hepatic tumours in rabbits and doubled the embolisation rate of microspheres in the tumour compartment (P < 0.05) compared to either norepinephrine or PRO alone [65].
The effect of PRO was also investigated in two human gastric carcinoma cell lines (SGC-7901 and BGC-823) by Liao and colleagues [66]. In vitro concentrations of 200 μM induced cell cycle arrest and apoptosis in both cell lines. The effect of PRO on the radiosensitivity of the same gastric carcinoma cell lines was also investigated [67]. When pre-treated for 24 hours with PRO at a concentration of 50 μM both cell lines displayed a significant increase in radiosensitivity and apoptosis. PRO was associated with a decreased level of NF-κB and down-regulation of VEGF, COX-2, and EGFR expression.
In non-small cell lung cancer (NSCLC), Al-Wadei and colleagues investigated the effects of chronic exposure to nicotine on cancer cell proliferation [68]. Results showed that nicotine-treated lung adenocarcinoma cells, (NCI-H322, NCI-H441 and NCI-H1299), released norepinephrine and increased proliferation. Cells treated with PRO at a concentration of 1 μM for 10-minutes prior to acute or chronic exposure to nicotine showed significant reductions in the number of viable cells compared to nicotine alone (P < 0.0001). Interestingly, exposure to PRO also reduced viability compared to cells not treated with nicotine. The same authors subsequently extended this line of research and showed a similar nicotine-drive autocrine catecholamine feedback loop in a pancreatic adenocarcinoma model [69].
Kozanoglu et al investigated the in vitro effect of PRO on the U266 human multiple myeloma cell line [70]. They showed a dose and time dependent effect on cell proliferation and apoptosis, with IC50 values of 141, 100, and 75 μM after 24-, 48-, and 72-h PRO exposure, respectively.
Abdi and colleagues used two doses of PRO, 5 mg/kg and 10 mg/kg, in mice bearing solid Ehrlich carcinoma tumours [71]. While both doses were associated with reductions tumour growth rates and volumes compared to untreated controls, it was only the higher dose group that was associated with improved survival (P < 0.05). Both dosage levels were associated with statistically significant reductions in IL-10, HSP70 and iNOS levels.
Wei et al explored the use of PRO in a panel of human thyroid cancer cell lines [72]. PRO was shown to inhibit growth of 8505C and K1 cell lines, with IC50 values of 200 μM and 280 μM, respectively. Growth inhibition was further analysed in 8505C line was shown to be associated with increased levels of apoptosis. In vivo data from a xenograft mouse model using 8505C cells and treated with PRO at a dose of 10 mg/kg showed reduced tumour volume increase compared to controls, with reduced SUVmax on PET/CT and Ki76 staining of tumour cells.
von Hippel-Lindau (VHL) disease, or von Hippel-Lindau syndrome, is a rare genetic disorder caused by a germ-line mutation in the von Hippel–Lindau tumour suppressor gene. It is characterised by the development of a range of tumour types, including hemangioblastoma of the retina and the central nervous system. Albiñana et al explored the effect of PRO on hemangioblastoma cells from VHL patients and reported a dose-dependent decrease in viability and increase in apoptosis [73]. This effect was enhanced in hypoxic conditions and was associated with a decreased the expression of HIF target genes, including VEGF.
Human Data
The earliest human data to suggest a positive effect of propranolol on cancer came from epidemiological studies comparing cancer incidence in hypertensive and non-hypertensive patients. For example Perron et al reported on the incidence of prostate cancer in Canadian men treated with a range of anti-hypertensive drugs and found that treatment with beta-blockers had a protective effect with an adjusted odds ratio (OR) of 0.86 (95% CI = 0.77– 0.96) [74]. In patients with hepatitis C associated cirrhosis Nkontchou et al reported that PRO use was associated with a decreased risk hepatocellular carcinoma (HCC), with a hazard ratio (HR) = 0.25; (95% CI = 0.09 – 0.65; P < 0.004) [75]. Chang et al published a large cohort study of over 24 238 patients, with a PRO group (> 6 months use, n = 12 119) compared to a non-PRO use group (n = 12 119) over a 12 year period [76]. Overall the risk of cancer was lower in the PRO group (HR = 0.75; 95% CI = 0.67–0.85; P < 0.001), and site specific analysis showed a decreased risk in head and neck cancers (HR = 0.58; 95% CI = 0.35–0.95), oesophagus (HR = 0.35; 95% CI = 0.13 – 0.96), stomach (HR = 0.54; 95% CI = 0.30 – 0.98), colon (HR = 0.68; 95% CI = 0.49 – 0.93), and prostate cancers (HR = 0.52; 95% CI = 0.33 – 0.83).
Zhong et al performed a systematic review and meta-analysis of observational studies of beta-blocker use and cancer mortality [77]. They identified 24 relevant studies that had reported results by May 2015, involving 76 538 patients. Post-diagnosis use of a beta-blocker was associated with a statistically signification reduction in the risk of all-cause mortality (HR = 0.89; 95% CI = 0.81 – 0.98; P = 0.02). When stratified by cancer type the reduction in risk of death was only significant for breast cancer (HR = 0.82; 95% CI = 0.68 – 0.99; P = 0.03). In terms of cancer-specific mortality, beta-blocker use was associated with a reduced risk of cancer-specific mortality (HR = 0.89; 95% CI = 0.79 – 0.99; P = 0.03). However on stratification by cancer type there was no beneficial effect of post-diagnosis use of beta-blockers for breast, colorectal or prostate cancer. For pre-diagnosis use of beta-blockers there was no significant effect on all-cause mortality, but stratification by cancer type showed both a benefit for melanoma (HR = 0.81; 95% CI = 0.67 – 0.97; P = 0.02) and an increased risk for ovarian cancer (HR = 1.17; 95% CI = 1.04 – 1.32; P = 0.01). However, Weberpals et al evaluated a possible immortal time bias in these observational studies and found no clinically meaningful evidence for an association between beta-blocker use and survival when restricting the analysis to studies not prone to immortal time bias. Careful interpretation of observational studies is therefore required when no attempt is made to address immortal time bias [78].
Breast Cancer
Results published by Barron et al in a larger population of breast cancer patients showed protective effects associated with PRO use [79]. Irish women treated with PRO (n = 70) or atenolol (n = 525) in the year prior to breast cancer diagnosis were matched with women not receiving beta-blocker treatment (n = 4738) in the ratio 1:2. PRO use was associated with a lower risk of presenting with a T4 (OR = 0.24, 95% CI = 0.07 – 0.85) or N2/N3/M1 (OR = 0.20; 95%C = 0.04 – 0.88) diagnosis compared to matched non-users. The cumulative probability of breast cancer-specific mortality was also significantly lower for PRO users compared to matched nonusers (HR = 0.19; 95% CI = 0.06 – 0.60). There was no difference in either outcome between atenolol users and matched non-users.
A systematic review and meta-analysis of beta-blocker use and breast cancer by Childers et al found a non-significant reduction in breast cancer recurrence (HR = 0.67; 95% CI = 0.39 - 1.13), a significant reduction in the risk of breast cancer mortality (HR = 0.50; 95% CI = 0.32 - 0.80) and no impact of all-cause mortality (HR = 1.02; 95% CI = 0.75 - 1.37) [80].
Angiosarcoma
Banavali and colleagues reported on a case of relapsing metastatic angiosarcoma treated with a combination of metronomic oral low-dose chemotherapy (etoposide and cyclophosphamide), celecoxib and PRO (40 mg twice a day) [81]. The patient showed a complete response after two cycles of therapy. After one year of treatment the patient remained on a maintenance treatment of oral cyclophosphamide (50 mg) and PRO (20 mg BID), on alternate days for six additional months after which treatment ceased. The patient relapsed 20 months after initiation of the metronomic treatment and was treated with local palliative radiotherapy and oral thalidomide 100 mg with some response but ultimately died of progressive disease.
Subsequently the same group have reported on a series of seven cases of advanced angiosarcoma treated with a combination of PRO (40 mg BID), weekly vinblastine (i.v. 6 mg/m2 to a maximum of 10 mg) and methotrexate (35 mg/m2 to a maximum 50 mg) for up to 12 months followed by maintenance of PRO (40 mg BID), oral etoposide (50 mg/day) and oral cyclophosphamide (50 mg/day) for 20 consecutive days in cycles of 30 days [44]. The treatment was well tolerated and showed a 100% response rate, including one complete response and three very good partial responses. Median PFS was 11 months (range 5–24) and OS was 16 months (range 10–30).
Chow et al also published a case report in angiosarcoma treated with PRO [82]. The patient presented with a widely disseminated non-metastatic multifocal stage T2 cutaneous angiosarcoma, a diagnosis with a 2-year survival rate of 0%. Treatment with PRO at a dose of 40 mg BID was initiated leading to clinical improvement within a week and a subsequent increase in the dose to 40 mg three times a day. Staining for Ki-67 showed that the PRO monotherapy was associated with a 34% reduction in proliferation rate. Subsequent treatments, administered concurrently with PRO, included 10-weeks of paclitaxel poliglumex (a formulation of paclitaxel designed to increase the therapeutic index of the drug) and radiotherapy. Following cessation of paclitaxel and radiotherapy the patient has continued to take PRO at a dose of 40 mg TID, with clear signs of disease regression and no evidence of metastatic disease.
Other
Bhattacharyya et al reported on the use of the combination of metronomic temozolomide (mTMZ), the COX-2 inhibitor etodolac and PRO in recurrent glioblastoma at ASCO 2014 [83]. A series of 32 patients were randomised to either mTMZ or mTMZ with VT-122 (the combination of PRO 20 mg BID and etodolac 400 mg BID). The median TTP was 5.2 months in the mTMZ arm and 8.8 months in the mTMZ VT-122 arm. Survival at six months was 40% and 63%, and OS at one year was 12% versus 22% respectively. An update in 2015, also reported at ASCO, with 41 patients included, showed a median OS of 9.2 months versus 17.6 months, and a response rate of 35% versus 57% respectively [84].
The same group also reported on a small (n = 37) single centre open-label trial of gemcitabine and nab-paclitxel (GemNab) with and without VT-122 in patients with locally advanced or metastatic pancreatic cancer [85]. Patients in the GemNab VT-122 arm (n = 20) were treated with VT-122 for one week prior to commencement of GemNab, and then continuously with GemNab. PFS was 7.2 and 11.8 months, and OS was 10.5 months versus 15.9 months for the GemNab and GemNab VT-122 arms respectively. Additionally patients in the GemNab VT-122 arm experienced reduced pain and neuropathy and increased weight gain compared to the GemNab patients.
VT-122 was also used in a small (n = 20), multi-centre, randomised controlled trial in combination with sorafenib in HCC [86]. Median OS was 9.6 months versus 17.2 months in the sorafenib and sorafenib VT-122 arms respectively.
There has also been some interest in the role of PRO in addressing cancer-related cachexia, a significant cause of morbidity and mortality in late-stage cancer patients [87]. Hyltander et al explored influences on resting energy expenditure (REE) in weight-losing cancer patients taking PRO (80 mg BID), the non-selective COX inhibitor indomethacin, morphine or placebo [88]. Patients in the PRO group showed a decrease in REE of around 10% compared to base-line after 5 days of treatment (P < 0.02), whereas there were no significant changes in the other treatment groups or controls. A subsequent study by the same group compared PRO (80 mg/day) and atenolol (50 mg/day) in 10 weight-losing solid tumour patients [89]. While both drugs significantly reduced REE (P < 0.05), PRO treatment, accounting for a decline in heart rate, was significantly more pronounced compared with atenolol (P<0.05).
There has also been some interest in the psychological effects of PRO in cancer patients, for example in reducing the level of emotional distress, measured in terms of the number and rate of intrusive thoughts, associated with a cancer diagnosis [90].
Clinical Trials
As of 31st May 2016, a number of clinical trials are investigating the anti-cancer uses of PRO. Only trials which are currently open (recruiting or soon to commence recruitment) or on-going are included.
Feasibility/Phase I
NCT01504126 – A small single arm, open-label (n = 25) trial of PRO, 20 mg BID, with surgery and standard platinum or taxane chemotherapy in ovarian cancer. This is designated as a feasibility study, with a primary end-point of the proportion of patients completing treatment. Treatment commences 48 – 72 hours prior to surgical resection and continues for six cycles of chemotherapy post-surgery.
NCT02013492 – A feasibility study of oral PRO in patients with non-resectable recurrent or metastatic solid tumours. Patients in this open-label study are treated with PRO BID (dose not specified) for four months in the absence of disease progression or unacceptable toxicity. Primary end-points are incidence of toxicity, change in VEGF levels, measurement of impacts on immune response and tumour microenvironment. Secondary outcome measures include one-year PFS and OS.
NCT02897986 (PROVIN) – This planned Phase I dose escalation trial (10, 20 and 30 mg/m² of thrice weekly oral vinorelbine only plus daily PRO 1.5mg/kg/day BID) after completion of the first cycle. Pharmacokinetic analysis of vinorelbine and PRO will be performed. Once the recommended dose of the combination is established four extension cohorts of 9 patients (neuroblastoma, rhabdomyosarcoma, Ewing’s sarcoma, and miscellaneous tumours) will be added. Potential biomarkers (tumour beta-adrenergic receptor expression and beta-tubulin isotypes) will also be evaluated.
NCT02732678 – A dose-finding trial of PRO in combination with metronomic fixed oral cyclophosphamide in patients with locally advanced or metastatic angiosarcoma (PROPAN)
Randomised – no phase listed
NCT00502684 – A randomised, placebo-controlled trial of peri-operative PRO and etodolac in women undergoing breast cancer surgery. Women in the treatment arm will receive 40 mg of PRO and 800 mg of etodolac starting two days prior to surgery and continuing until three days post-surgery. The trial has a number of biochemical primary outcome measures including immune and angiogenesis-related biomarkers (number and cytotoxic activity of NK cells, levels of NK T-cells, lymphocytes, monocytes and granulocytes; levels of cortisol and VEGF). The primary clinical end-point is the five-year recurrence rate.
Phase II
NCT01847001 – A Phase II open-label study in newly diagnosed breast cancer patients undergoing neo-adjuvant chemotherapy. The starting dose of PRO is 20 mg BID, but is up-titrated to 40 mg then 80 mg BID. Primary outcomes are PRO compliance during chemotherapy, changes in angiogenesis (as assessed using Diffuse Optical Tomography) and changes in psychological stress levels. Secondary outcomes include adverse event rates and changes in tumour proliferation (Ki-67 staining).
NCT02596867 – A Phase II open-label ‘window of opportunity’ trial in newly diagnosed breast cancer. PRO, at a dose of 1.5 mg/kg BID, is administered for three weeks prior to surgical resection. The primary outcome is a reduction in the proliferative index (Ki-67), secondary outcomes relate to safety, toxicity and adherence.
NCT01988831 – A Phase II randomised, placebo controlled trial in high-risk primary melanoma. Patients at high risk of disease recurrence will receive PRO at a dose assessed by a cardiologist, to a maximum of 160 mg/day. The primary outcome is five-year PFS. [Recruitment currently suspended due to financial reasons].
NCT01857817 – A Phase II randomised, placebo controlled trial of PRO and etodolac in prostate cancer. The drug combination, at a specific dose of 22 mg PRO and 340 mg etodolac, is designated as VT-122, by the sponsor of the trial, Vicus Therapeutics. The dose used in this trial is 22 mg PRO and 340 mg of etodolac twice a day. Primary outcome is change in PSA at 12 weeks. Secondary outcomes include PSA doubling time, PSA progression and time to symptom progression.
NCT01265576 – A Phase II randomised, placebo-controlled trial of VT-122 with sorafenib in hepatocellular carcinoma (HCC) patients at risk of cachexia. The primary outcome is failure-free survival at 6-months. The clinical benefit rate at 6-months is the secondary outcome. Note that a previous trial of VT-122 in NSCLC-related cachexia has yet to report results (trial completed December 2012).
NCT02641314 – A Phase II randomised trial of metronomic chemotherapy and PRO in children and adolescents with recurrent or progressive high risk neuroblastoma (METRO-NB2012). Treatment consists of eight alternating 28-day-cycles of PRO, celecoxib, oral cyclophosphamide, fortnightly i.v. vinblastine, oral etoposide (PCCVE) and of PRO, celecoxib, oral cyclophosphamide, fortnightly i.v. vinblastine (PCCV) followed by five cycles PCCV resulting in a total of 13 cycles (364 days of treatment). The daily dose of PRO is 0.5 mg/kg/day, to a maximum of 120 mg/day, in two divided doses. The primary outcome is to demonstrate the non-inferiority of event free survival (EFS) in comparison to a historical control group. Secondary outcomes include the disease control rate at 6 months and overall survival at 12 months.
ACTRN12615000889550 – A Phase II randomised study of perioperative PRO vs placebo on gene expression in newly diagnosed breast cancer. The treatment group will receive 7 days of pre-operative PRO (40 mg BID days 1 – 3, 80 mg BID days 4 - 8) prior to and including the day of surgery and then will be titrated off PRO over two days in the post-operative period. The primary outcome is tumour gene expression for each of 20,000 genes at baseline and at surgical resection.
ACTRN12612000852853 – A Phase II randomised controlled trial of peri-operative PRO and etodolac in colorectal cancer patients undergoing surgical excision of the primary tumour. The primary outcome is a reduction of two-year rate of recurrence and distant metastases.
Phase III
NCT00888797 – This is a sister trial to NCT00502684 and is a Phase III randomised, placebo-controlled trial of peri-operative PRO and etodolac in colorectal cancer patients undergoing resection. Patients in the treatment arm receive etodolac 800 mg BID for the entire intervention period, PRO 20 mg BID for 5 pre-operative days, 80 mg BID on the day of surgery, 40 mg BID for the first postoperative week, 20 mg PO BID for the second postoperative week. The primary clinical end-point is the rate of local and distant recurrence rate at three years. Secondary end points are immune-related.
EudraCT 2014-003671-30 – This open label Phase III trial in patients with VHL syndrome will assess the efficacy of PRO, at a dose of 2 mg/kg, in controlling the growth of papillary and juxta-papillary retinal hemangioblastomas. The primary endpoint is a reduction in the number and size of retinal hemangioblastomas at 12-months.
Mechanisms of Action
PRO is a non-selective beta-adrenergic receptor antagonist, with a similar binding affinity for beta1- and beta2- and a much lower affinity (approximately 100-fold) for the beta3-adrenoreceptor [91]. In this respect PRO has a similar selectivity to some other clinically used beta-blocker drugs, particularly pindolol and carvedilol and there exists some evidence that these particular drugs may also have potential in repurposing [92–93]. There are a number of distinct putative mechanisms of action that have been investigated in relation to the anticancer effects of PRO, many of them associated with the beta2-adrenoreceptor pathway and which may be particularly important in the context of the metastatic process [94].
Proliferation
Investigations of the influence of beta-adrenergic signalling on cellular proliferation extend back more than fifty years. In 1961 Selye, Veilleux and Cantin reported that rats chronically treated with the beta-adrenergic agonist isoproterenol displayed excessive growth of salivary glands, most likely due to an increased mitotic rate [95]. Barka subsequently confirmed that isoproterenol increased the rate of mitosis and triggered DNA synthesis [96]. The converse, a reduction of proliferation due to PRO was also reported in the same era, for example Iwata, Kariya and Fujimoto showed that in a ciliated protozoan beta-adrenergic blockade, with PRO and other agents, was associated with a reduced rate of growth [97]. It has since been shown that PRO is able to inhibit the increase in cancer cell proliferation associated with catecholamines or isoproterenol in a number of cancer cell types [56, 98–100].
In a comparison between PRO and the selective beta adrenergic receptor antagonists atenolol (beta1) and ICI118-551 (beta 2), Işeri and colleagues showed that PRO and ICI118,551 were more potent in reducing the proliferation, migration, and invasion of non-stimulated breast (MCF7), colon (HT-29), and hepatocellular (HepG2) cancer cells than atenolol [101].
In contrast, there have also been contradictory results, for example for breast cancer where Pérez Piñero et al have reported that beta adrenergic agonists isoprenaline and salbutamol reduced breast tumour growth in animal models, and that PRO treatment reversed this inhibition [102] and other authors have reported a decrease in proliferation in breast cancer cell line, for example in the MCF7 cell line [101].
Bernabé et al showed that OSCC proliferation in response to increased beta2 adrenergic signalling was mediated by IL-6 [61]. In addition to showing increased IL-6 production from OSCC cells lines in response to norepinephrine or isoproterenol, neutralising IL-6 using antibodies inhibited the increase in proliferation rate. PRO was also shown to inhibit the increased proliferation rate induced by IL-6. A decrease in circulating IL-6 levels in response to PRO has been reported in a murine melanoma model, concomitant with a lower level of metastatic growth compared to untreated controls [103].
Migration and Invasion
Strell et al have shown that PRO is able to abrogate the norepinephrine-induced increase in migratory activity of a range of breast cancer cell lines [28–29].
Degradation of the extra-cellular matrix is a key factor in tumour progression and the metastatic cascade, with the matrix metalloproteinases (MMPs) playing a central role in the tissue remodelling process [104–105]. Sood et al reported that MMP-2 and MMP-9 were highly expressed during the norepinephrine-induced increase in ovarian cancer invasiveness [39]. Furthermore they showed that PRO treatment could reduce the rate of tumour growth and infiltration in vivo. A similar inhibition of the upregulation of MMP-2 and MMP-9 by PRO has also been reported in nasopharyngeal carcinoma [59], pancreatic cancer [50, 53], gastric adenocarcinoma [66], melanoma [106], prostate cancer [107] and of MMP-9 in medulloblastoma [108] and infantile hemangioma [109–110].
Pon et al described a beta2 adrenergic receptor activated feed-forward loop driving the invasiveness of the highly metastatic MDA-MB-231-HM breast cancer cell line [111]. A range of beta2 adrenergic receptor agonists caused a dose dependent increase in cAMP and Ca2 signalling and a decrease in phosphorylated ERK, which was competitively inhibited by PRO and the selective beta2 adrenergic receptor antagonist ICI-118551. The increased invasion of the MDA-MB-231-HM cell line was shown to be associated with the positive feedback between cAMP and Ca2 but independent of the effect on pERK.
Tissue remodelling is one of the key steps in the process of epithelial-mesenchymal transition (EMT) which is important in metastasis [112]. Shan et al showed that the human gastric cancer cell lines BGC-823 and SGC-7901 underwent morphological changes typical of EMT, and showed increased invasiveness, in response to co-culture with norepinephrine via a beta2-adrenergic signalling pathway [113]. Zhang et al showed a similar association between increased catecholamine signalling and initiation of EMT in HT-29 and A549 colorectal cancer cell lines [114]. Additionally they showed that TGF-β1 mediated the norepinephrine induced EMT process and that PRO, at a concentration of 10 μM, inhibited the increase in TGF-β1 and partially decreased the migration and invasiveness of norepinephrine-treated cells.
Related to the degradation of the extra-cellular matrix and the EMT process is the formation of tumour cell invadopodia, which are actin structures formed on the surface of cancer cells believed to be active in the secretion of MMPs [115]. Creed et al have shown that beta2 adrenergic signalling increased the rate of invadopodia formation in human and murine breast cancer cell lines in vitro, and that PRO inhibited this increase [116].
Also important in the metastatic process is cell-cell adhesion, and here too there is evidence that adrenergic signalling is active. Rap1B is a small GTPase that increases cell-cell adhesion. There is evidence to suggest that loss of Rap1B at the plasma membrane decreases cell-cell adhesion and may promote a metastatic phenotype [117–118]. MDA-MB-231 breast cancer cells showed reduced cell-cell adhesion in response to isoproterenol and an increase in cell migration, whereas treatment with PRO reduced the level of migration [119].
The process of metastasis depends also on the creation of a ‘metastatic niche’, in addition to the properties of the primary cancer cells [120–121]. Campbell at al showed that in a mouse model restraint-induced stress or exogenous isoproterenol promoted MDA-231 breast cancer cell colonization of bone via adrenergic signalling effects on the bone marrow stromal compartment [31]. This effect was via upregulation of RANKL, which increased the number of osteolytic lesions in response to catecholamine signalling, and that this effect could be reversed by PRO or the RANKL inhibitor denosumab.
The role of the lymphatic system in metastasis has become increasingly recognised in recent years [122]. Le et al showed that chronic stress induced a remodelling of the tumour lymphatic vasculature, including an increase in lymphatic vessel density and vessel dilation leading to elevated levels of lymphatic draining [123]. These effects were sensitive to beta-adrenergic signalling and could be enhanced by isoproterenol or inhibited by PRO treatment of BALB/c mice injected with MDA-MB-231 breast cancer cells. The stress-induced remodelling was mediated by VEGFC production, which was induced by COX-2/PGE2. PRO has also been explored as a treatment option for congenital lymphangioma [124].
Apoptosis
Zhang et al showed that PRO limited the expansion of the PC-2 pancreatic cancer cell line and that this was due to an increased rate of apoptosis [51]. The pro-apoptotic action of PRO was found to be via blockade of the beta2-adrenergic receptor rather than beta1, as shown by the increased level of apoptosis induced by the selective beta2 antagonist butaxamine and the reduced level due to the beta1 blocker metoprolol. Chin and colleagues also showed that in colorectal cancer cell lines PRO was associated with cell cycle arrest and apoptosis [57].
Liao et al reported that in vitro PRO concentrations of 200 μM induced cell cycle arrest and apoptosis in gastric carcinoma cell lines. Apoptosis was associated with a decrease in levels of NF-κB, VEGF, COX-2, MMP-2 and MMP-9 expression [66].
The pro-apoptotic activity of PRO was also investigated by Wolter and colleagues in their work in head and neck squamous cell carcinoma (HNSCC) cell lines with differing p53 status [62]. PRO treatment was shown to cause apoptosis irrespective of p53 status and was related to down-stream activity of p63 and p73, both p53-family proteins. Following PRO treatment there was evidence of downregulation of the anti-apoptotic ΔNp63a and induction of the pro-apoptotic TAp73b in both SCC9 and SCC17a cell lines. Some of the same authors also investigated the pro-apoptotic effect of PRO on neuroblastoma cell lines and showed that treatment increased expression of p53 and p73 [47].
Angiogenesis
The relationship between adrenergic signalling and angiogenesis was first elucidated in the late 1990s, when it was shown that beta adrenergic signalling by norepinephrine induced increased levels of VEGF expression in brown adipose tissue [125–126].
Lutgendorf and colleagues showed that beta adrenergic agonists increased the expression of VEGF in two ovarian cancer cell lines (EC and SKOV3), and that PRO, at a concentration of 1 μM, blocked this increase [38]. This finding suggested a putative link between behavioural stress and enhanced tumour growth via increased angiogenesis. In vivo work using a murine model of ovarian cancer showed that chronic behavioural stress was associated with increased tumour growth and vascularisation and enhanced expression of VEGF, MMP-2 and MMP-9. In particular beta-adrenergic activation of the cAMP-PKA signalling pathway was identified as a major mechanism by which behavioural stress enhanced tumour angiogenesis [127]. In subsequent work some of the same authors showed that surgical stress induced increased beta-adrenergic signalling in a murine ovarian cancer model and that this was associated with increased rates of tumour growth and tumour angiogenesis [40]. Perioperative PRO was shown to inhibit the surgical stress-induced increase in VEGF expression and the consequent increase in angiogenesis and tumour growth.
An anti-angiogenic effect of PRO, via down-regulation of VEGF has also been shown in a range of cancer cell lines including nasopharyngeal carcinoma [59], melanoma [128], pancreatic cancer [50], leukaemia [23], head and neck squamous cell carcinoma [62] and infantile hemangiomas [129–130]. Pasquier et al showed that the combination of beta-blockers, including PRO, with vincristine was associated with increased survival and reduced angiogenesis in a mouse model of neuroblastoma [45].
Other mechanisms may also play a role in the anti-angiogenic effects of PRO. For example, Annabi et al, following initial reports that PRO was effective in infantile hemangiomas [131], investigated PRO activity in human glioblastoma biopsy samples [60]. It was reported that PRO down-regulated endothelial MMP-9 expression and reduced the rate of human brain microvascular endothelial cells tubologenesis, potentially reducing tumour angiogenesis.
Park et al showed that hypoxia-inducible factor 1α (HIF-1α) expression is also upregulated by norepinephrine, via the cAMP/PKA/Akt/p70S6K pathway, in addition to VEGF, and that it plays a key role in the angiogenic process [132]. Silencing of HIF-1α reduced the norepinephrine-induced increase in VEGF expression and capillary tube formation. Furthermore PRO pre-treatment abrogated the effect of adrenergic signalling on HIF-1α, VEGF and angiogenesis. Similar results, using a beta2-adrenergic receptor antagonist (ICI118 551), have been shown in vitro and in vivo in a murine pancreatic cancer model [133].
In addition to VEGF, MMP-2 and MMP-9, Liao et al showed that isoproterenol increased levels of COX-2, in gastric cancer cell lines, while PRO significantly reduced expression (P < 0.05) [66]. The COX-2/prostaglandin E2 (PGE2) pathway is also known to be involved in cancer-associated angiogenesis [134–135]. These effects were partly due to activation of the activation of NF-κB pathway [66]. Ciccarelli and colleagues showed that genetic deletion of b2-adrenergic receptors impaired angiogenesis in a mouse model, and that isoproterenol induced IκBα degradation and enhanced NF-kB transcriptional activity in a time-dependent manner [136].
Treatment Sensitisation
Early in vitro work showed that PRO had the potential to revert the drug resistant phenotype in different cell lines, including doxorubicin-resistant P388 murine leukaemia [18] and human multi-drug resistant (MDR) CEM leukaemia [137] for example. But results were cell line and drug specific, for example PRO seemed to have little impact on the cisplatin sensitivity of NSCLC cell lines [138].
In addition to chemosensitisation there has also been some preclinical work investigating the relationship between PRO and radiation. Liao et al showed that pre-treatment of the human gastric adenocarcinoma (HGC) cell lines BGC-823 and SGC-7901 with PRO, at a concentration of 50 μM for 24 hours, increased the effect of radiotherapy on cell viability in vitro [67]. Similarly, Wolter et al assessed the impact of PRO on HNSCC cell lines and showed that it enhanced the effect of radiation, in addition to displaying evidence of synergy with cisplatin [62]. We may also note the concurrent use of PRO and chemoradiotherapy in one of the case reports of PRO use in angiosarcoma [82].
The mechanism for increased radiosensitivity may be related to the NF-κB/COX-2/PGE2 pathway inhibition that a number of investigators have reported [36, 52, 67]. Evidence exists to suggest that elevated COX-2 expression may confer increased radiation resistance in some cancer cell lines [139–141].
Pasquier et al investigated the synergism of PRO with paclitaxel and 5-FU, both in vitro and in vivo [30]. In vitro analysis of a number of human cancer and non-cancer lines showed a range of synergistic, additive, sub-additive and antagonistic effects on cell proliferation depending on dose, cell line and chemotherapy drug. The synergistic effects were shown to be due to an enhancement of the anti-angiogenic effects of the chemotherapy drugs by low concentrations (10 μM) of PRO. In vivo a murine orthotopic triple negative breast cancer (MDA-MB-231) xenograft model was used with each chemotherapy drug. Four treatment groups of tumour-bearing mice were used, control (saline-treatment), paclitaxel alone (20 mg/kg, 3 days a week for 3 weeks), PRO alone (10 mg/kg, 5 days a week for 5 weeks) or the combination of paclitaxel and PRO. The same protocol was used with 5-FU, and the dose of the drug was 5-FU alone (30 mg/kg, 3 days a week for 5 weeks). The combination treatments produced significantly improved median survival times both for paclitaxel (125 days vs 70 for paclitaxel alone, or 47 for control, P = 0.0005) and 5-FU (56 days vs 47 for 5-FU or 44 for control, P = 0.0005). Subsequently the same group published results which showed PRO synergised with vincristine in a murine model of neuroblastoma [45].
There are still other examples of PRO acting to improve cancer cell sensitivity to drug treatment enhancing the effect of rapamycin on human prostate cancer PC3 cells [49], reverting resistance to trastuzumab in HER2 breast cancer [32], inhibiting the stress-related reduction of sunitinib activity in colorectal cancer in an in vivo model [142] and sensitising thyroid cancer cells to the targeted BRAF-V600E inhibitor vemurafenib [72].
Immunological
A number of immune-related mechanisms of action have also been outlined as important in the anti-cancer effects of PRO. These effects are primarily mediated by the effects of sympathetic nervous system (SNS) signalling on different populations of immune cells, the tumour microenvironment and, in some cases, directly on cancer cells. An important aspect of this complex relationship is the link between psychological stress and immune response, a key concern within the field of psychoneuroimmunology and increasingly important in the context of cancer [143]. While there are a number of papers which review the effect of physical and psychological stress on the immune system, for example [144–145], the primary focus in this paper is on direct evidence of the role of PRO, and where relevant, other beta adrenergic receptor antagonists.
Teshima et al showed that in C3H/H and AKR mice PRO, at a dose of 5 mg/kg, was able to increase the phagocytic activity of macrophages (P < 0.05) and inhibit the reduction of phagocytic activity induced by physical restraint-induced stress (P <0.01) [146].
Shakhar and Ben-Eliyahu showed that the beta-adrenergic agonist metaproterenol induced a dose-dependent transient increase in natural killer (NK) cell numbers within 10 minutes of administration in F344 rats (P < 0.0001) [24]. It should be noted that although it did not reach significance, the time course of NK numbers showed that the initial increase subsided within one hour and before falling below base-line values by three hours and returning to base-line at five hours. Blood NK activity was depressed by metaproterenol (P < 0.03), but this was inhibited by prior administration of PRO (at doses in the range 0.1 – 0.5 mg/kg) or nadolol. Additional experiments showed that nadolol, like PRO a non-selective beta-adrenergic receptor antagonist, reduced the number of lung-retained NK-sensitive MADB106 breast cancer cells and the number of lung metastases.
Subsequently Benish et al showed that surgical intervention (laparotomy) prior to inoculation with MADB106 cells in F344 rats was significantly (P < 0.05) associated with increased the rate of lung tumour cell retention (LTR) [25]. This increase in LTR was attenuated by pre-surgical treatment with COX-2 inhibitors (indomethacin, etodolac, and celecoxib). Pre-surgical PRO, at a dose of 1.5 mg/kg and 4.5 mg/kg, also significantly reduced the LTR rate compared to untreated controls. The combined treatment of etodolac (12.5 mg/kg) and PRO (1.5 mg/kg) was more effective than either single treatment and completely inhibited the effect of surgery. Notably the combined treatment reversed the surgically induced reduction in NK cell numbers and per-cell cytotoxicity. These deleterious impacts on NK cells were later shown to be associated with reduced FasL and CD11a expression post-surgery, and that the combined etodolac PRO treatment counter-acted these effects [36].
Kalinichenko et al investigated the effect of norepinephrine on cytotoxic T lymphocytes (CTL) using a MOPC-315 plasmocytoma model in BALB/c mice [147]. Results showed that norepinephrine inhibited CTL generation via a reduction of TNF-α expression in CD4 and CD8 T cells and F4/80 activated macrophages, a result confirmed by the addition of exogenous TNF-α. Ex vivo use of PRO, at a concentration of 1 μM, completely reversed the effects of norepinephrine on TNF-α and CTL generation. Sloan et al also identified stress-sensitive CD11b F4/80 macrophages as being implicated in the metastatic process in the same mouse model [27].
Wu et al showed that stress induced by social isolation in Balb/c mice injected with colon 26-L5 carcinoma cells increased the rate of liver metastases compared to unstressed controls (P < 0.05) [148]. This was shown to be associated with a reduction in thymus weight, reduction in thymocytes and reduction in cytolytic activity of NK cells. Subsequent research by the same laboratory replicated these findings in additional mouse models and also showed that over-crowding induced similar stress-related thymic atrophy [33]. Additionally it was shown that PRO, at a dose of 30 ppm, slowed the rate of tumour growth of the over-crowded group to below that of the unstressed controls.
Kanemi et al showed that stressed induced by physical restraint in C57BL/6 mice resulted in a significant increase in epinephrine levels (P < 0.001) which returned to base-line values within one hour [149]. Lymphocyte numbers in blood and lungs were depressed (P <0.001) by restraint, but numbers returned to base-line within four hours of cessation of restraint. Lung NK cell numbers were also reduced by restraint-induced stress (P < 0.01), as were all other lymphocyte subsets assessed (CD8 , CD4 , B cell and NKT cells). PRO, at a dose of 20 mg/kg, administered prior to restraint was shown to reverse the reduction in NK cell numbers compared to untreated controls but had no effect on other lymphocyte sub-sets in the lungs.
In contrast, Tarr et al showed that repeated social disruption stress (induced by repeatedly introducing aggressive mice into cages of non-aggressive mice), both increased splenic NK cell numbers, NK cytotoxic activity and the expression of activation markers, both immediately and 14 hours after stress [150]. Administration of PRO (10 mg/kg) reduced the ‘priming’ of these NK cells at 14 hours. The authors propose an evolutionary explanation for these findings, suggesting that the priming occurs in order to prepare the host for pathogenic insult during stressful ‘fight or flight’ episodes. However, it has been argued that the complexity of multiple immune compartments, NK cell lineages and complexity of following cell populations in vivo over extended time periods makes interpretation of these results difficult [151].
Catecholamines are also known to impact the immune response via down-regulation of interferon gamma (IFN-γ) production [152]. Khalili et al showed that PRO and a HSP-70-rich tumour lysate vaccine synergised to increase IFN-γ production in a murine model of fibrosarcoma [153]. Treated animals showed lower rates of tumour growth (P < 0.01) and increased level of CTL activity (P < 0.05).
Lymphocytes are also known to secrete catecholamines, with potential downstream impacts on immunity. Huang et al investigated the effect of lymphocyte-derived catecholamines on the differentiation and function of T helper (Th) cells, suggesting that they shifted the Th1/Th2 balance in the direction of greater Th2 polarisation [154–155]. Panina-Bordignon et al had earlier suggested that beta2-adrenergic signalling inhibits production of IL-12, thereby promoting Th2 differentiation and inhibiting the Th1 development associated with anti-tumour immunity [156].
Myeloid derived suppressor cells (MDSC) are implicated in the dysfunctional immune response to cancer and are considered a negative prognostic marker in some cancers [157–158]. Jin et al used BALB/c mice to show that chronic stress induced an immunosuppressive state associated with an accumulation of CD11b Gr1 MDSCs in the bone marrow [159]. In line with previous reports from other groups [160], the data showed that the COX-2/PGE2 axis a central role in this accumulation. In addition there was evidence that stress-related catecholamines were also implicated, and that PRO (10mg/kg) partially reversed the accumulation of MDSCs, both in terms of cell numbers (P < 0.001) and proportion (P = 0.018).
T-regulatory (T-reg) cells are another population of immune cells associated with tumour-associated immune dysfunction. Zhou et al studied the impact of PRO and the COX-2 inhibitor parecoxib on T-reg numbers in breast cancer patients undergoing radical mastectomy [161]. Patients were assigned to control, PRO, parecoxib and PRO parecoxib groups. Patients in the PRO group received 20 mg TID starting from day of surgery until third post-operative day. The parecoxib group received 40 mg per day, IV, from day of surgery to second post-operative day. Patients in the combination group received both treatments at the same dose and schedule. Results showed that surgery was associated with an increase in epinephrine, norepinephrine, PGE2 levels and T-reg numbers. Treatment with PRO or PRO parecoxib attenuated the increase in T-reg numbers though there was no additive or synergistic effect of the parecoxib compared to PRO alone. Ex vivo analysis showed that PRO also reduced the immunosuppressive effect of T-reg cells compared to controls.
Other
In addition to the main mechanisms that have been outlined about, there are also a number of other possible mechanisms of action which have been described in the literature, some of which may be unrelated to the beta-adrenergic receptor antagonist activity of PRO.
Epidermal growth factor receptor (EGFR) signalling plays a central role in many cancer types and is a major drug target [162]. Disrupted endocytic trafficking is implicated in the process by which tumours gain self-sufficiency in growth signals by delays in the inactivation of multiple growth factor receptors, including EGFR [163–164]. It has been proposed by Shaughnessy et al that a strategy for inhibiting EGFR function may be to interfere with the endocytic process directly rather than directly targeting receptor-ligand binding or tyrosine kinase activity [165]. Inhibition of phosphatidic acid phosphohydrolase (PAP) has been shown to cause a reversible trafficking of inactive (empty) EGFR from the cell surface to endosomes, thereby restricting receptor availability to ligands [166]. PRO is a known inhibitor of PAP [167], and has been shown to reduce the cell viability of EGFR-dependent cancer cell lines [165].
Kang et al investigated the impact of PRO on glucose uptake in breast cancer cell lines (4T1, MDA-MB-231 and MCF-7) [168]. In vitro analysis showed that while glucose transporter-1 (GLUT-1) was relatively unaffected in all cell lines by PRO, hexokinase-2 (HK-2) was sensitive to PRO at a concentration of 50 μM. In vivo results using the 4T1 mouse breast cancer line showed that tumours were also sensitive to PRO at a dose of 10 mg/kg, and that this was associated with a reduction in HK-2 expression. PRO may therefore have a metabolic impact on tumour growth.
PGE2 is a key inflammatory molecule with multiple effects in cancer, including effects on immunity, angiogenesis, proliferation and apoptosis [169]. As has already been shown, these diverse effects are also implicated in some of the mechanisms of action of PRO. Nagaraja et al sought to investigate the relationship between beta adrenergic signalling and PGE2 in more detail using primary ovarian cancer cells from patients [170]. Tumour samples from depressed patients showed significantly higher levels of PGE2 and PGF2α, tumour samples from mice bearing Skov3-ip1 ovarian tumours also showed elevated levels of PGE2. Analysis showed that norepinephrine increased the secretion of PGE2 from tumour cells. Furthermore PRO was also shown to inhibit the catecholamine-induced increase in PGE2 in Skov3 and HeyA8 ovarian cancer cells.
Platelets play a complex role in tumour progression and metastasis via the release of pro-angiogenic factors, a role in subverting anti-tumour immunity by ‘cloaking’ tumour cells from NK cells and a role in establishing the metastatic niche [171]. The anticancer effects of aspirin may be related to anti-platelet effects via irreversible inhibition of COX-1 [172–173], and there is also some evidence that other anti-platelet agents, such as low molecular weight heparins and dipyridamole may also have anti-cancer or anti-metastatic activity [174–176]. Beta-adrenergic receptor antagonists are also known to have effects on platelet aggregation and a meta-analysis published in 2014 showed that they decreased platelet aggregation by 13% (95% CI = 8 - 17%, standardised mean difference=-0.54, 95% CI = -0.85 – -0.24, P < 0.0001) [177]. In particular non-selective lipophilic beta-blockers (including PRO) decreased platelet aggregation more than selective non-lipophilic beta-blockers. A small randomised cross-over trial in moderate essential hypertension compared PRO (40mg TID) to atenolol (100 mg/day) confirmed that the number of circulating platelet aggregates decreased significantly with PRO (0.99±0.19) in comparison with both atenolol (1.41±0.70; P = 0.004) and baseline (1.59±0.94; P = 0.002) [178].
Finally, beta-adrenergic signalling is at the intersection between psychological states and physiology. As has been previously mentioned stress arising from social interactions has been shown to have negative effects on proliferation, invasion, metastasis and anticancer immunity. However, it should be noted that not all forms of social stress are necessarily negative – clinical psychology differentiates between positive stress (eustress) and negative stress (distress) with differing physiological correlates [179]. Cao et al showed that a murine model of eustress, related to living in an enriched environment (increased levels of inanimate stimulation, social stimulation and physical exercise), was associated with reduced rate of B16 melanoma growth compared living in a control environment [180]. This reduction in tumour growth rate was associated with down-regulation of hypothalamic brain-derived neurotrophic factor (BDNF) and increased production of leptin in adipocytes via beta-adrenergic signalling. PRO, at a dose of 0.5 g/l in drinking water, inhibited the protective effect of the enriched environment.
Our Take
The evidence outlined above, and summarised in Table 1, suggests that PRO has a number of distinct anti-cancer effects which may be of therapeutic value in different clinical settings. While there is evidence that other drugs in the same class as have anti-cancer effects, for example carvedilol and nebivolol [45], PRO has both the widest range of evidence and extensive clinical use, therefore pragmatic reasons suggest that it be prioritised for further clinical research. Needless to say, the repurposing of other beta-blockers may also carry great potential in the same or other settings/tumours. For PRO the evidence in angiosarcoma, anti-metastatic effects in breast and ovarian cancers and the effect on the rate of distant metastases following surgical intervention are particularly noteworthy. In all future trials, it will be important to assess plasma concentrations of propranolol especially when a direct effect on tumour growth is desirable.
Angiosarcoma
Angiosarcoma is a rare and aggressive soft tissue sarcoma arising in vascular endothelial cells, and is a disease with poor prognosis and reported five-year overall survival rates in the range 30% - 40% [181]. Although no standard treatment exists, the majority of patients are treated with surgical resection, chemotherapy (doxorubicin or paclitaxel most commonly) and radiotherapy [182]. Given the high unmet needs in this disease, the case reports showing some benefit to patients using PRO in combination with other agents are noteworthy [44, 81, 82], along with responses to propranolol observed in other vascular tumours [41, 183–184]. Clinical trials are urgently required to confirm the efficacy of these combinations with PRO.
Anti-metastatic Agent
Metastatic disease remains the primary cause of cancer-related mortality and therefore the search for anti-metastatic agents is of considerable value [185]. As outlined previously, PRO has multiple mechanisms of action which may have some impact on the metastatic process, including reduction in the rate of invasion, down-regulation of angiogenesis and lymphangiogenesis, inhibition of tissue remodelling and down-regulation of COX-2/PGE2 expression. Data from a number of in vivo models has shown that PRO may reduce the rate of metastasis in breast and other cancers via direct effects on beta-adrenergic signalling [27, 36, 48]. The addition of PRO to standard of care for non-metastatic cancers may therefore be a strategy to reduce the rate of metastatic spread.
Table 1. Summary of evidence by cancer type.
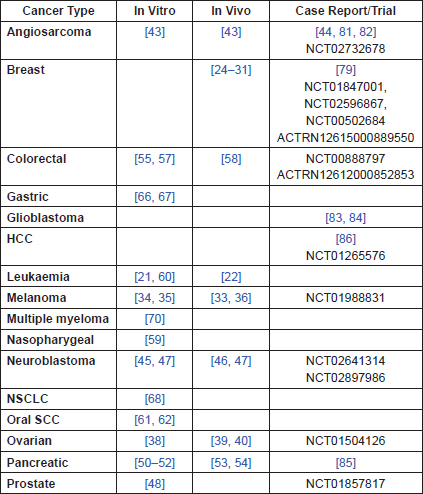
Perioperative Intervention
Data from both retrospective analyses of patient outcomes and from animal models suggest that surgical intervention may be associated with distant metastases [186–187]. The ‘wound healing response’ due to the surgical incision initiates a cascade of inflammatory events that lead to suppression of cell-mediated immunity and an increase in pro-angiogenic signalling [188–191]. However, there is now a growing level of interest in targeting some aspects of this post-surgical response so as to reduce the risk of metastatic spread [192–193]. Options for such peri-operative interventions include choice of anaesthesia [194–195], the use of drugs which target the COX-2/PGE2 pathway such as ketorolac [196–198] and diclofenac [199–200], and the H2RA cimetidine [201–202]. As should be clear from the results previously outlined, beta-adrenergic signalling is also implicated in the post-surgical metastatic process and numerous in vivo studies have reported that peri-operative PRO is associated with a reduced rate of metastases [40]. Of note the combination of PRO with a COX-2/PGE2 inhibitor, such as ketorolac or etodolac, has the potential to show synergism in a peri-operative setting and warrants additional investigation. This is particularly the case in those cancers in which post-surgical distant metastases are a frequent occurrence, including breast cancer, osteosarcoma, head and neck cancers, upper GI cancers, NSCLC and ovarian cancer.
Other Cancers
While we have outlined areas where there is a particularly compelling case to be made for further clinical study, there is also sufficient evidence to suggest that there may be some value in assessing the potential benefit of PRO in other cancers. The ubiquity of catecholamine signalling and the apparent expression of adrenergic receptors in multiple tumour types would suggest that the effects of PRO may extend, ultimately, to a much larger number of cancer types than has hitherto been suggested [203–204]. In the meantime there is a good level of evidence for further investigation in pancreatic cancer and neuroblastoma, particularly in combination with other agents, including repurposed drugs such as ketorolac or etodolac.
Psychological Stress
We note that the data for a pro-apoptotic effect of PRO on primary tumour growth comes primarily from in vitro studies which use high concentrations which may not reflect physiologically achievable levels. In many respects this is similar to the case with some NSAIDs, such as diclofenac, where the evidence is that the apoptotic effects are not physiologically achievable, and that therefore the anti-cancer effects are related to aspects of the host environment rather than directly on primary cancer growth [205]. Beta adrenergic signalling is central to the intersection between psychosocial stress and cancer, as evidenced in numerous animal models and epidemiological data. A variety of stress reduction techniques have been clinically investigated, including mindfulness meditation [206], cognitive behavioural stress management [207] and even communal singing [208]. Therefore, while the data for an effect on primary tumour growth may be limited in contrast to the results supporting an anti-metastatic effect, there is reason to believe that beta blockade may be beneficial in terms of psychological effects in addition to physical effects on the host environment.
Next Steps
The current level of clinical trial activity, which is relatively high for a repurposed drug, testifies to the strong level of clinical evidence and it is to be hoped that positive reports from these trials will be forthcoming in the future.
The data are strongest for clinical trials of PRO, in combination with other agents, in the following cancer types:
• Angiosarcoma
• Breast cancer
• Ovarian cancer
• Pancreatic cancer
• Neuroblastoma
The peri-operative use of PRO in combination with ketorolac or etodolac is also of interest in the following cancers:
• Osteosarcoma
• Head and neck cancers
• Oesophageal cancer
• Breast cancer
• Ovarian cancer
• Non-small Cell Lung Cancer
• Pancreatic cancer
Conclusion
There is a significant volume of data from in vitro, animal and human studies to indicate that there are multiple clinically relevant anti-cancer effects associated with PRO. This data has been summarised and presented to make the case that PRO is a very strong candidate for repurposing as an anticancer agent. In particular the potential for synergistic interactions with other drugs has been outlined, including repurposed COX-2/PGE2 inhibitors and a range of chemotherapeutics at both metronomic and standard dosing. The anti-metastatic properties of PRO may be particularly valuable to exploit during surgical intervention, and a number of possible combinations with other agents is discussed in this setting.
Author Contributions
Primary author: Pan Pantziarka. Contributing authors: Ilse Rooman, Vidula Sukhatme, Gauthier Bouche, Lydie Meheus, Vikas P. Sukhatme. All authors read and approved the final manuscript.
Competing Interests
The authors declare that they have no competing interests. All the authors are associated with not for profit organisations that aim to repurpose drugs for oncology treatments. VPS is also a scientific advisory board member of Berg Health and Mitra Biotech and a consultant to Roivant Sciences.
References
1. Ali A et al (2015) Propranolol attenuates hemorrhage and accelerates wound healing in severely burned adults Crit Care 19 217 DOI: 10.1186/s13054-015-0913-x PMID: 25936635 PMCID: 4432824
2. Peskind ER et al (2005) Propranolol for disruptive behaviors in nursing home residents with probable or possible Alzheimer disease: a placebo-controlled study Alzheimer Dis Assoc Disord 19(1) 23–8 DOI: 10.1097/01.wad.0000155067.16313.5e PMID: 15764868
3. Greendyke RM et al (1986) Propranolol treatment of assaultive patients with organic brain disease. A double-blind crossover, placebo-controlled study J Nerv and Ment Dis 174(5) 290–4 DOI: 10.1097/00005053-198605000-00005
4. Joint_Formulary_Committee (2015) British National Formulary 69th ed BMJ Group and Pharmaceutical Press
5. Fogari R et al (1990) Beta-blocker effects on plasma lipids in antihypertensive therapy: importance of the duration of treatment and the lipid status before treatment J Cardiovasc Pharmacol 16(Suppl 5) S76–80 DOI: 10.1097/00005344-199006165-00013 PMID: 11527140
6. McEvoy GK (2004) Propranolol American Society Health-System Pharmacists 1777–84
7. Shand DG (1976) Pharmacokinetics of propranolol: a review Postgrad Med J 52(Suppl 4) 22–5 PMID: 787953
8. Walle T et al (1981) Food-induced increase in propranolol bioavailability–relationship to protein and effects on metabolites Clin Pharmacol Ther 30(6) 790–5 DOI: 10.1038/clpt.1981.239 PMID: 7307427
9. Bowman SL et al (1986) A comparison of the pharmacokinetics of propranolol in obese and normal volunteers. Br J Clinical Pharmacol 21(5) 529–32 DOI: 10.1111/j.1365-2125.1986.tb02837.x
10. Leahey WJ et al (1980) Comparison of the efficacy and pharmacokinetics of conventional propranolol and a long acting preparation of propranolol Bri J Cclin Pharmacol 9(1) 33–40 DOI: 10.1111/j.1365-2125.1980.tb04793.x
11. Olesen J, Hougård K and Hertz M (n.d.) Isoproterenol and propranolol: ability to cross the blood-brain barrier and effects on cerebral circulation in man Stroke 9(4) 344–9 DOI: 10.1161/01.STR.9.4.344
12. Filippi L et al (2016) Successful propranolol treatment of a kaposiform hemangioendothelioma apparently resistant to propranolol PediatrBlood Cancer 63(7) 1290–2
13. Mutschler E, Spahn H and Kirch W (1984) The interaction between H2-receptor antagonists and beta-adrenoceptor blockers Bri J Clin Pharmacol 17 Suppl 1 51S–7S DOI: 10.1111/j.1365-2125.1984.tb02428.x
14. Winniford MD, Huxley RL and Hillis LD (1983) Randomized, double-blind comparison of propranolol alone and a propranolol-verapamil combination in patients with severe angina of effort J Am Coll Cardiol 1(2 Pt 1) 492–8 DOI: 10.1016/S0735-1097(83)80078-3 PMID: 6338082
15. Martin TJ et al (1978) Characterization of beta-adrenergic receptor linked to adenylate cyclase in a human cancer cell line (COLO 16) Clin Sci Mol Med 55(1) 23–9 PMID: 27327
16. Schmitt H and Pochet R (1977) In vivo labelling of beta-adrenergic receptors on rat glioma cells FEBS Lett 76(2) 302–5 DOI: 10.1016/0014-5793(77)80173-7 PMID: 16780
17. Delavier-Klutchko C, Hoebeke J and Strosberg a D (1984) The human carcinoma cell line A431 possesses large numbers of functional beta-adrenergic receptors FEBS Lett 169(2) 151–5 DOI: 10.1016/0014-5793(84)80308-7 PMID: 6143691
18. Ramu A et al (1984) Restoration of doxorubicin responsiveness in doxorubicin-resistant P388 murine leukaemia cells Brit J Cancer 50(4) 501–7 DOI: 10.1038/bjc.1984.207 PMID: 6487516 PMCID: 1976913
19. Tsuruo T et al (1984) Effects of quinidine and related compounds on cytotoxicity and cellular accumulation of vincristine and adriamycin in drug-resistant tumor cells Cancer Res 44(10) 4303–7 PMID: 6467192
20. Shiraishi N et al (1986) Lysosomotropic agents reverse multiple drug resistance in human cancer cells Cancer Lett 30(3) 251–9 DOI: 10.1016/0304-3835(86)90049-2
21. Hajighasemi F and Mirshafiey A (2009) In vitro sensitivity of leukemia cells to propranolol. J Clin Med Res 1(3) 144–9 PMID: 22493648 PMCID: 3318877
22. Lamkin DM et al (2012) Chronic stress enhances progression of acute lymphoblastic leukemia via β-adrenergic signaling Brain Behav Immun 26(4) 635–41 DOI: 10.1016/j.bbi.2012.01.013 PMID: 22306453 PMCID: 3322262
23. Hajighasemi F and Hajighasemi S (2009) Effect of propranolol on angiogenic factors in human hematopoietic cell lines in vitro Iran Biomed J 13(4) 223–8 PMID: 19946348
24. Shakhar G and Ben-Eliyahu S (1998) In vivo beta-adrenergic stimulation suppresses natural killer activity and compromises resistance to tumor metastasis in rats J Immunol 160(7) 3251–8 PMID: 9531281
25. Benish M et al (2008) Perioperative use of beta-blockers and COX-2 inhibitors may improve immune competence and reduce the risk of tumor metastasis Ann Surg Oncol 15(7) 2042–52 DOI: 10.1245/s10434-008-9890-5 PMID: 18398660 PMCID: 3872002
26. Goldfarb Y et al (2011) Improving postoperative immune status and resistance to cancer metastasis: a combined perioperative approach of immunostimulation and prevention of excessive surgical stress responses Ann Surg 253(4) 798–810 DOI: 10.1097/SLA.0b013e318211d7b5 PMID: 21475023
27. Sloan EK et al (2010) The sympathetic nervous system induces a metastatic switch in primary breast cancer Cancer Res 70(18) 7042–52 DOI: 10.1158/0008-5472.CAN-10-0522 PMID: 20823155 PMCID: 2940980
28. Lang K et al (2004) Induction of a metastatogenic tumor cell type by neurotransmitters and its pharmacological inhibition by established drugs Int J Cancer 112(2) 231–8 DOI: 10.1002/ijc.20410 PMID: 15352035
29. Strell C et al (2012) Norepinephrine promotes the β1-integrin-mediated adhesion of MDA-MB-231 cells to vascular endothelium by the induction of a GROα release Mol Cancer Res 10(2) 197–207 DOI: 10.1158/1541-7786.MCR-11-0130
30. Pasquier E et al (2011) Propranolol potentiates the anti-angiogenic effects and anti-tumor efficacy of chemotherapy agents: implication in breast cancer treatment Oncotarget 2(10) 797–809 DOI: 10.18632/oncotarget.343 PMID: 22006582 PMCID: 3248157
31. Campbell JP et al (2012) Stimulation of host bone marrow stromal cells by sympathetic nerves promotes breast cancer bone metastasis in mice PLoS biology 10(7) e1001363 DOI: 10.1371/journal.pbio.1001363 PMID: 22815651 PMCID: 3398959
32. Liu D et al (2015) β2-AR signaling controls trastuzumab resistance-dependent pathway. Oncogene 35(1) 1–12
33. Hasegawa H and Saiki I (2002) Psychosocial stress augments tumor development through beta-adrenergic activation in mice Jpn J Cancer Res 93(7) 729–35 DOI: 10.1111/j.1349-7006.2002.tb01313.x PMID: 12149137
34. Dal Monte M et al (2013) Functional involvement of β3-adrenergic receptors in melanoma growth and vascularization J Mol Med (Berl) 91(12) 1407–19 DOI: 10.1007/s00109-013-1073-6
35. Wrobel LJ and Le Gal FA (2014) Inhibition of human melanoma growth by a non-cardioselective β-Blocker J Invest Dermatol 135(2) 1–7
36. Glasner A et al (2010) Improving survival rates in two models of spontaneous postoperative metastasis in mice by combined administration of a b-adrenergic antagonist and a cyclooxygenase-2 Inhibitor J Immunol 184(5) 2449–57 DOI: 10.4049/jimmunol.0903301 PMID: 20124103
37. Lutgendorf SK et al (2002) Vascular endothelial growth factor and social support in patients with ovarian carcinoma Cancer 95(4) 808–15 DOI: 10.1002/cncr.10739
38. Lutgendorf SK et al (2003) Stress-related mediators stimulate vascular endothelial growth factor secretion by two ovarian cancer cell lines Clinical Cancer Res 9(12) 4514–21
39. Sood AK et al (2006) Stress hormone-mediated invasion of ovarian cancer cells Clin Cancer Research 12(2) 369–75 DOI: 10.1158/1078-0432.CCR-05-1698
40. Lee J et al (2009) Surgical stress promotes tumor growth in ovarian carcinoma Clin Cancer Res 15(8) 2695–702 DOI: 10.1158/1078-0432.CCR-08-2966 PMID: 19351748 PMCID: 2746852
41. Léauté-Labrèze C et al (2015) A randomized, controlled trial of oral propranolol in infantile hemangioma New Engl J Med 372(8) 735–46 DOI: 10.1056/NEJMoa1404710 PMID: 25693013
42. Chisholm KM et al (2012) β-Adrenergic receptor expression in vascular tumors. Mod Pathol 25(11) 1446–51 DOI: 10.1038/modpathol.2012.108 PMID: 22743651
43. Stiles JM et al (2013) Targeting of beta adrenergic receptors results in therapeutic efficacy against models of hemangioendothelioma and angiosarcoma PloS One 8(3) e60021 DOI: 10.1371/journal.pone.0060021 PMID: 23555867 PMCID: 3610939
44. Pasquier E et al (2016) Effective management of advanced angiosarcoma by the synergistic combination of propranolol and vinblastine-based metronomic chemotherapy: a bench to bedside study EBioMedicine 6 87–95 DOI: 10.1016/j.ebiom.2016.02.026 PMID: 27211551 PMCID: 4856748
45. Pasquier E et al (2013) β-blockers increase response to chemotherapy via direct antitumour and anti-angiogenic mechanisms in neuroblastoma Br J Cancer 108(12) 2485–94 DOI: 10.1038/bjc.2013.205 PMID: 23695022 PMCID: 3694229
46. Xu T et al (2013) Antiangiogenic effect of propranolol on the growth of the neuroblastoma xenografts in nude mice J Pediatr Surg 48(12) 2460–5 DOI: 10.1016/j.jpedsurg.2013.08.022 PMID: 24314187
47. Wolter JK et al (2014) Anti-tumor activity of the beta-adrenergic receptor antagonist propranolol in neuroblastoma Oncotarget 5(1) 161–72 PMID: 24389287 PMCID: 3960198
48. Palm D et al (2006) The norepinephrine-driven metastasis development of PC-3 human prostate cancer cells in BALB/c nude mice is inhibited by beta-blockers Int J Cancer 118(11) 2744–9 DOI: 10.1002/ijc.21723
49. Brohée L et al (2015) Lipin-1 regulates cancer cell phenotype and is a potential target to potentiate rapamycin treatment Oncotarget 6(13) 11264–80 DOI: 10.18632/oncotarget.3595 PMID: 25834103 PMCID: 4484455
50. Guo K et al (2009) Norepinephrine-induced invasion by pancreatic cancer cells is inhibited by propranolol Oncol Rep 22(4) 825–30 PMID: 19724861
51. Zhang D et al (2009) Inhibition of pancreatic cancer cell proliferation by propranolol occurs through apoptosis induction: the study of beta-adrenoceptor antagonist’s anticancer effect in pancreatic cancer cell Pancreas 38(1) 94–100 DOI: 10.1097/MPA.0b013e318184f50c
52. Zhang D et al (2010) β2-adrenergic antagonists suppress pancreatic cancer cell invasion by inhibiting CREB, NFκB and AP-1 Cancer Biol Ther 10(1) 19–29 DOI: 10.4161/cbt.10.1.11944 PMID: 20424515
53. Kim-Fuchs C et al (2014) Chronic stress accelerates pancreatic cancer growth and invasion: a critical role for beta-adrenergic signaling in the pancreatic microenvironment Brain Behav Immun 40 40–7 DOI: 10.1016/j.bbi.2014.02.019 PMID: 24650449 PMCID: 4102665
54. Partecke LI et al (2016) Chronic stress increases experimental pancreatic cancer growth, reduces survival and can be antagonised by beta-adrenergic receptor blockade Pancreatology 16(3) 423–33 DOI: 10.1016/j.pan.2016.03.005 PMID: 27083074
55. Masur K et al (2001) Norepinephrine-induced migration of SW 480 colon carcinoma cells is inhibited by beta-blockers Cancer Res 61(7) 2866–9 PMID: 11306460
56. Coelho M et al (2015) Antiproliferative effects of β-blockers on human colorectal cancer cells Oncol Rep 33(5) 2513–20 PMID: 25812650
57. Chin C-C et al (2016) Selective β2-AR blockage suppresses colorectal cancer growth through regulation of EGFR-Akt/ERK1/2 signaling, G1-phase arrest and apoptosis J Cell Physiol 231(2) 459–72 DOI: 10.1002/jcp.25092
58. Lin Q et al (2013) Effect of chronic restraint stress on human colorectal carcinoma growth in mice PloS One 8(4) e61435 DOI: 10.1371/journal.pone.0061435 PMCID: 3621827
59. Yang EV et al (2006) Norepinephrine up-regulates the expression of vascular endothelial growth factor, matrix metalloproteinase (MMP)-2, and MMP-9 in nasopharyngeal carcinoma tumor cells Cancer Res 66(21) 10357–64 DOI: 10.1158/0008-5472.CAN-06-2496 PMID: 17079456
60. Annabi B et al (2009) Propranolol adrenergic blockade inhibits human brain endothelial cells tubulogenesis and matrix metalloproteinase-9 secretion Pharmacological Res 60(5) 438–45 DOI: 10.1016/j.phrs.2009.05.005
61. Bernabé DG et al (2011) Stress hormones increase cell proliferation and regulates interleukin-6 secretion in human oral squamous cell carcinoma cells Brain Behav Immun 25(3) 574–83 DOI: 10.1016/j.bbi.2010.12.012
62. Wolter NE et al (2012) Propranolol as a novel adjunctive treatment for head and neck squamous cell carcinoma J Otolaryngol Head Neck Surg 41(5) 334–44 PMID: 23092836
63. Grzanna R et al (1985) In vivo administration of propranolol delays development of a murine plasmacytoma tumor J Auton Nerv Syst 14(4) 397–401 DOI: 10.1016/0165-1838(85)90085-2 PMID: 4086728
64. Bomber P, McCready R and Hammersley P (1986) Propranolol hydrochloride enhancement of tumor perfusion and uptake of gallium-67 in a mouse sarcoma J Nucl Med 27(2) 243–5 PMID: 3712042
65. Burton M and Gray BN (1987) Redistribution of blood flow in experimental hepatic tumours with noradrenaline and propranolol Br J Cancer 56(5) 585–8 DOI: 10.1038/bjc.1987.245 PMID: 3426921 PMCID: 2001891
66. Liao X et al (2010) The β-adrenoceptor antagonist, propranolol, induces human gastric cancer cell apoptosis and cell cycle arrest via inhibiting nuclear factor κB signaling Oncol Rep 24(6) 1669–76 PMID: 21042766
67. Liao X et al (2010) Effects of propranolol in combination with radiation on apoptosis and survival of gastric cancer cells in vitro Radiat Oncol (London, England) 5 98 DOI: 10.1186/1748-717X-5-98
68. Al-Wadei HA, Al-Wadei MH and Schuller HM (2012) Cooperative regulation of non-small cell lung carcinoma by nicotinic and beta-adrenergic receptors: a novel target for intervention PLoS.One 7(1) e29915 DOI: 10.1371/journal.pone.0029915
69. Al-Wadei MH, Al-Wadei HAN and Schuller HM (2012) Pancreatic cancer cells and normal pancreatic duct epithelial cells express an autocrine catecholamine loop that is activated by nicotinic acetylcholine receptors α3, α5, and α7 Mol Cancer Res 10(2) 239–49 DOI: 10.1158/1541-7786.MCR-11-0332 PMCID: 3340883
70. Kozanoglu I et al (2013) New indication for therapeutic potential of an old well-known drug (propranolol) for multiple myeloma J Cancer Res Clin Oncol 139(2) 327–35 DOI: 10.1007/s00432-012-1331-y
71. Abdin AA, Soliman NA and Saied EM (2014) Effect of propranolol on IL-10, visfatin, Hsp70, iNOS, TLR2, and survivin in amelioration of tumor progression and survival in Solid Ehrlich Carcinoma-bearing mice Pharmacol Rep 66(6) 1114–21 DOI: 10.1016/j.pharep.2014.07.010 PMID: 25443743
72. Wei W-J et al (2016) Propranolol sensitizes thyroid cancer cells to cytotoxic effect of vemurafenib Oncol Rep 36(3) 1576–84 PMID: 27432558
73. Albiñana V et al (2015) Propranolol reduces viability and induces apoptosis in hemangioblastoma cells from von Hippel-Lindau patients Orphanet J Rare Dis 10(1) 118 DOI: 10.1186/s13023-015-0343-5 PMID: 26394686 PMCID: 4579575
74. Perron L et al (2004) Antihypertensive drug use and the risk of prostate cancer (Canada) Cancer Causes Control 15(6) 535–41 DOI: 10.1023/B:CACO.0000036152.58271.5e PMID: 15280632
75. Nkontchou G et al (2012) Effect of long-term propranolol treatment on hepatocellular carcinoma incidence in patients with HCV-associated cirrhosis Cancer Prev Res (Philadelphia, Pa.) 5(8) 1007–14 DOI: 10.1158/1940-6207.CAPR-11-0450
76. Chang P-Y et al (2015) Propranolol reduces cancer risk: a population-based cohort study Medicine(Baltimore) 94(27) p. e1097
77. Zhong S et al (2015) β-Blocker use and mortality in cancer patients: systematic review and meta-analysis of observational studies Eur J Cancer Prev 25(5) 440–8 DOI: 10.1097/CEJ.0000000000000192 PMID: 26340056
78. Weberpals J et al (2016) Beta blockers and cancer prognosis - The role of immortal time bias: A systematic review and meta-analysis Cancer Treat Rev 47 1–11 DOI: 10.1016/j.ctrv.2016.04.004 PMID: 27179912
79. Barron TI et al (2011) Beta blockers and breast cancer mortality: a population- based study J Clin Oncol 29(19) 2635–44 DOI: 10.1200/JCO.2010.33.5422 PMID: 21632503
80. Childers WK, Hollenbeak CS and Cheriyath P (2015) β-Blockers Reduce Breast Cancer Recurrence and Breast Cancer Death: A Meta-Analysis Clin Breast Cancer 15(6) 426–31 DOI: 10.1016/j.clbc.2015.07.001 PMID: 26516037
81. Banavali S, Pasquier E and Andre N (2015) Targeted therapy with propranolol and metronomic chemotherapy combination: sustained complete response of a relapsing metastatic angiosarcoma Ecancermedicalscience 9 499 DOI: 10.3332/ecancer.2015.499 PMID: 25624880 PMCID: 4303616
82. Chow W et al (2015) Growth attenuation of cutaneous angiosarcoma with propranolol-mediated β-blockade JAMA Dermatol 151(11) 1226–9 DOI: 10.1001/jamadermatol.2015.2554 PMID: 26375166
83. Bhattacharyya GS et al (2014) Continuous metronomic temozolamide with propranolol and etodolac in recurrent globlastoma: A pilot study in ASCO Annual Meeting Proceedings e13005
84. Shahid T et al (2015) Continuous low dose temozolamide with or without VT-122 in glioblastoma in ASCO Annual Meeting Proceedings e13010
85. Bhattacharyya GS et al (2015) Effect of coadministered beta blocker and COX-2 inhibitor to patients with pancreatic cancer prior to receiving albumin-bound (Nab) paclitaxel in ASCO Annual Meeting Proceedings
86. de la Torre AN et al (2015) Effect of coadministration of propranolol and etodolac (VT-122) plus sorafenib for patients with advanced hepatocellular carcinoma (HCC) J Clin Oncol 33(suppl 3; abstr 390)
87. Aoyagi T et al (2015) Cancer cachexia, mechanism and treatment World J Gastrointest Oncol 7(4) 17–29 PMID: 25897346 PMCID: 4398892
88. Hyltander A, Körner U and Lundholm KG (1993) Evaluation of mechanisms behind elevated energy expenditure in cancer patients with solid tumours Euro J Clin Invest 23(1) 46–52 DOI: 10.1111/j.1365-2362.1993.tb00716.x
89. Hyltander A et al (2000) Beta-adrenoceptor activity and resting energy metabolism in weight losing cancer patients EurJ Cancer 36(3) 330–4 DOI: 10.1016/S0959-8049(99)00273-7
90. Lindgren ME et al (2013) Beta-blockers may reduce intrusive thoughts in newly diagnosed cancer patients Psychooncology 22(8) 1889–94 DOI: 10.1002/pon.3233 PMCID: 3612565
91. Hoffmann C et al (2004) Comparative pharmacology of human beta-adrenergic receptor subtypes–characterization of stably transfected receptors in CHO cells Naunyn-Schmiedebergs Arch Pharmacol 369(2) 151–9 DOI: 10.1007/s00210-003-0860-y PMID: 14730417
92. Qinye F et al (2014) Carvedilol suppresses migration and invasion of malignant breast cells by inactivating Src involving cAMP/PKA and PKCδ signaling pathway J Cancer Res Ther 10(4) 998–1003 DOI: 10.4103/0973-1482.137664
93. Lin C-S et al (2015) Carvedilol use is associated with reduced cancer risk: A nationwide population-based cohort study Int J Cardiol 184 9–13 DOI: 10.1016/j.ijcard.2015.02.015 PMID: 25705003
94. Cole SW et al (2015) Sympathetic nervous system regulation of the tumour microenvironment Nat Rev Cancer 15(9) 563–72 DOI: 10.1038/nrc3978 PMID: 26299593 PMCID: 4828959
95. Selye H, Veilleux R and Cantin M (1961) Excessive stimulation of salivary gland growth by isoproterenol Science 133(3445) 44–5 DOI: 10.1126/science.133.3445.44 PMID: 13749973
96. BARKA T (1965) Induced cell proliferation: the effect of isoproterenol Exp Cell Res 37(1865) 662–79 DOI: 10.1016/0014-4827(65)90214-4 PMID: 14315088
97. Iwata H, Kariya K and Fujimoto S (1969) Effect of compounds affecting the adrenergic mechanism on cell growth and division of Tetrahymena pyriformis W Jpn J Pharmacol 19(2) 275–81 DOI: 10.1254/jjp.19.275 PMID: 4391852
98. Bijman JT et al (1987) Modulation of placental alkaline phosphatase activity and cytokeratins in human HN-1 cells by butyrate, retinoic acid, catecholamines and histamine Br J Cancer 56(2) 127–32 DOI: 10.1038/bjc.1987.169 PMID: 2444242 PMCID: 2002146
99. Schuller HM and Cole B (1989) Regulation of cell proliferation by beta-adrenergic receptors in a human lung adenocarcinoma cell line Carcinogenesis 10(9) 1753–5 DOI: 10.1093/carcin/10.9.1753 PMID: 2569945
100. Lin X et al (2012) Beta-adrenoceptor action on pancreatic cancer cell proliferation and tumor growth in mice Hepatogastroenterology 59(114) 584–8 PMID: 22353526
101. Işeri OD et al (2014) beta-Adrenoreceptor antagonists reduce cancer cell proliferation, invasion, and migration Pharm Biol 52(11) 1374–81 DOI: 10.3109/13880209.2014.892513 PMID: 25026350
102. Pérez Piñero C et al (2012) Involvement of α2- and β2-adrenoceptors on breast cancer cell proliferation and tumour growth regulation Br J Pharmacol 166(2) 721–36 DOI: 10.1111/j.1476-5381.2011.01791.x PMCID: 3417500
103. Valles SL et al (2013) Stress hormones promote growth of B16-F10 melanoma metastases: an interleukin 6- and glutathione-dependent mechanism J Transl Med 11 72 DOI: 10.1186/1479-5876-11-72 PMID: 23517603 PMCID: 3608962
104. Shay G, Lynch CC and Fingleton B (2015) Moving targets: Emerging roles for MMPs in cancer progression and metastasis Matrix Biol 44–6 200–6 DOI: 10.1016/j.matbio.2015.01.019
105. Yadav L et al (2014) Matrix metalloproteinases and cancer - roles in threat and therapy. Asian Pac J Cancer Prev 15(3) 1085–91 DOI: 10.7314/APJCP.2014.15.3.1085 PMID: 24606423
106. Moretti S et al (2013) β-adrenoceptors are upregulated in human melanoma and their activation releases pro-tumorigenic cytokines and metalloproteases in melanoma cell lines Lab Invest 93(3) 279–90 DOI: 10.1038/labinvest.2012.175 PMID: 23318885
107. Barbieri A et al (2015) The stress hormone norepinephrine increases migration of prostate cancer cells in vitro and in vivo Int J Oncol 47(2) 527–34 PMID: 26058426
108. Annabi B et al (2010) Pharmacological targeting of β-adrenergic receptor functions abrogates NF-κB signaling and MMP-9 secretion in medulloblastoma cells Onco Targets Ther 3 219–26 DOI: 10.2147/OTT.S14503 PMID: 21124749 PMCID: 2994204
109. Wu S et al (2015) Clinical efficacy of propranolol in the treatment of hemangioma and changes in serum VEGF, bFGF and MMP-9 Exp Ther Medicine 10(3) 1079–83
110. Thaivalappil S et al (2013) Propranolol-mediated attenuation of MMP-9 excretion in infants with hemangiomas JAMA Otolaryngol Head Neck Surg 139(10) 1026–31 DOI: 10.1001/jamaoto.2013.4773 PMID: 24135743
111. Pon CK et al (2016) The β2-adrenoceptor activates a positive cAMP-calcium feedforward loop to drive breast cancer cell invasion FASEB J 30(3) 1144–54 DOI: 10.1096/fj.15-277798
112. Smith BN and Bhowmick NA (2016) Role of EMT in metastasis and therapy resistance J Clin Med 5(2) 17 DOI: 10.3390/jcm5020017 PMCID: 4773773
113. Shan T et al (2014) Novel regulatory program for norepinephrine-induced epithelial-mesenchymal transition in gastric adenocarcinoma cell lines Cancer Sci 105(7)847–56 DOI: 10.1111/cas.12438 PMID: 24815301 PMCID: 4317918
114. Zhang J et al (2016) Norepinephrine induced epithelial-mesenchymal transition in HT-29 and A549 cells in vitro J Cancer Res Clin Oncol 142(2) 423–35 DOI: 10.1007/s00432-015-2044-9
115. Jacob A and Prekeris R (2015) The regulation of MMP targeting to invadopodia during cancer metastasis Front Cell Dev Biol 3 4 DOI: 10.3389/fcell.2015.00004 PMID: 25699257 PMCID: 4313772
116. Creed SJ et al (2015) β2-adrenoceptor signaling regulates invadopodia formation to enhance tumor cell invasion Breast Cancer Res 17(1) 145 DOI: 10.1186/s13058-015-0655-3 PMID: 26607426 PMCID: 4660629
117. Linden J (2013) Adenosine promotes tumor metastasis Sci Signal 6(277) pe20 DOI: 10.1126/scisignal.2004290 PMID: 23716715
118. Ntantie E et al (2013) An adenosine-mediated signaling pathway suppresses prenylation of the GTPase Rap1B and promotes cell scattering Sci Signal 6(277) ra39 DOI: 10.1126/scisignal.2003374 PMID: 23716716 PMCID: 4278376
119. Wilson JM et al (2015) β-Adrenergic receptors suppress Rap1B prenylation and promote the metastatic phenotype in breast cancer cells Cancer Biol Ther 16(9) 1364–74 DOI: 10.1080/15384047.2015.1070988 PMID: 26209110 PMCID: 4622671
120. Psaila B and Lyden D (2009) The metastatic niche: adapting the foreign soil Nat Rev Cancer 9(4) 285–93 DOI: 10.1038/nrc2621 PMID: 19308068 PMCID: 3682494
121. Barcellos-Hoff MH, Lyden D and Wang TC (2013) The evolution of the cancer niche during multistage carcinogenesis Nat Rev Cancer 13(7) 511–8 DOI: 10.1038/nrc3536 PMID: 23760023
122. Karaman S and Detmar M (2014) Mechanisms of lymphatic metastasis J Clin Invest 124(3) 922–8 DOI: 10.1172/JCI71606 PMID: 24590277 PMCID: 3938272
123. Le CP et al (2016) Chronic stress in mice remodels lymph vasculature to promote tumour cell dissemination Nat Commun 7 10634 DOI: 10.1038/ncomms10634 PMID: 26925549 PMCID: 4773495
124. Ozeki M et al (2013) Propranolol as an alternative treatment option for pediatric lymphatic malformation Tohoku J Exp Med 229(1) 61–6 DOI: 10.1620/tjem.229.61
125. Asano A et al (1997) Adrenergic activation of vascular endothelial growth factor mRNA expression in rat brown adipose tissue: implication in cold-induced angiogenesis Biochem J 328(1) 179–83 DOI: 10.1042/bj3280179
126. Tonello C et al (1999) Role of sympathetic activity in controlling the expression of vascular endothelial growth factor in brown fat cells of lean and genetically obese rats FEBS Lett 442(2–3) 167–72 DOI: 10.1016/S0014-5793(98)01627-5 PMID: 9928995
127. Thaker PH et al (2006) Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma Nat Med 12(8) 939–44 DOI: 10.1038/nm1447 PMID: 16862152
128. Yang EV et al (2009) Norepinephrine upregulates VEGF, IL-8, and IL-6 expression in human melanoma tumor cell lines: implications for stress-related enhancement of tumor progression Brain Behav Immun 23(2) 267–75 DOI: 10.1016/j.bbi.2008.10.005 PMCID: 2652747
129. Lamy S et al (2010) Propranolol suppresses angiogenesis in vitro: inhibition of proliferation, migration, and differentiation of endothelial cells Vascul Pharmacol 53(5–6) 200–8 DOI: 10.1016/j.vph.2010.08.002 PMID: 20732454
130. Kum JJ and Khan ZA (2015) Mechanisms of propranolol action in infantile hemangioma Dermato-endocrinology 6(1) e979699 DOI: 10.4161/19381980.2014.979699 PMCID: 4580045
131. Léauté-Labrèze C et al (2008) Propranolol for severe hemangiomas of infancy New Engl J Med 358(24) 2649–51 DOI: 10.1056/NEJMc0708819 PMID: 18550886
132. Park SY et al (2011) Norepinephrine induces VEGF expression and angiogenesis by a hypoxia-inducible factor-1α protein-dependent mechanism Int J Cancer 128(10) 2306–16 DOI: 10.1002/ijc.25589
133. Shan T et al (2013) β2-AR-HIF-1α: a novel regulatory axis for stress-induced pancreatic tumor growth and angiogenesis Curr Mol Med 13(6) 1023–34 DOI: 10.2174/15665240113139990055 PMID: 23745588 PMCID: 4104615
134. Muraki C et al (2012) Cyclooxygenase-2 inhibition causes antiangiogenic effects on tumor endothelial and vascular progenitor cells Int J Cancer 130(1) 59–70 DOI: 10.1002/ijc.25976
135. Iñiguez MA et al (2003) Cyclooxygenase-2: a therapeutic target in angiogenesis Trends Mol Med 9(2) 73–8 DOI: 10.1016/S1471-4914(02)00011-4 PMID: 12615041
136. Ciccarelli M et al (2011) Impaired neoangiogenesis in β2-adrenoceptor gene-deficient mice: restoration by intravascular human β2-adrenoceptor gene transfer and role of NFκB and CREB transcription factors Bri J Pharmacol 162(3) 712–21 DOI: 10.1111/j.1476-5381.2010.01078.x
137. Zamora JM, Pearce HL and Beck WT (1988) Physical-chemical properties shared by compounds that modulate multidrug resistance in human leukemic cells Mol Pharmacol 33(4) 454–62 PMID: 3162758
138. Bando T et al (1997) Stimulation of beta-adrenoceptor enhances sensitivity to cisplatin in non-small cell lung cancer cell lines Int J Ooncol 10(6) 1197–201
139. Akutsu Y et al (2011) COX2 expression predicts resistance to chemoradiotherapy in esophageal squamous cell carcinoma Ann Surg Oncol 18(10) 2946–51 DOI: 10.1245/s10434-011-1645-z PMID: 21437756
140. Xia S et al (2010) Activated PI3K/Akt/COX-2 pathway induces resistance to radiation in human cervical cancer HeLa cells Cancer Biother Radiopharm 25(3) 317–23 DOI: 10.1089/cbr.2009.0707 PMID: 20578837
141. Yang HJ et al (2013) Investigation of radiation-induced transcriptome profile of radioresistant non-small cell lung cancer A549 cells using RNA-seq PloS One 8(3) e59319 DOI: 10.1371/journal.pone.0059319 PMID: 23533613 PMCID: 3606344
142. Liu J et al (2015) The effect of chronic stress on anti-angiogenesis of sunitinib in colorectal cancer models Psychoneuroendocrinology 52 130–42 DOI: 10.1016/j.psyneuen.2014.11.008
143. Green McDonald P, O’Connell M and Lutgendorf SK (2013) Psychoneuroimmunology and cancer: A decade of discovery, paradigm shifts, and methodological innovations Brain Behav Immun 30(Suppl) S1–9 DOI: 10.1016/j.bbi.2013.01.003 PMID: 23333846 PMCID: 3907949
144. Segerstrom SC and Miller GE (2004) Psychological stress and the human immune system: a meta-analytic study of 30 years of inquiry Psychol Bull 130(4) 601–30 DOI: 10.1037/0033-2909.130.4.601 PMID: 15250815 PMCID: 1361287
145. Glaser R and Kiecolt-Glaser JK (2005) Stress-induced immune dysfunction: implications for health Nat Rev Immunol 5(3) 243–51 DOI: 10.1038/nri1571 PMID: 15738954
146. Teshima H et al (1990) Prevention of immunosuppression in stressed mice by neurotropin(NSP) Life Sci 47(10) 869–76 DOI: 10.1016/0024-3205(90)90600-V PMID: 2215070
147. Kalinichenko VV et al (1999) Norepinephrine-mediated inhibition of antitumor cytotoxic T lymphocyte generation involves a beta-adrenergic receptor mechanism and decreased TNF-alpha gene expression J Immunol 163(5) 2492–9 PMID: 10452985
148. Wu W et al (2000) Social isolation stress enhanced liver metastasis of murine colon 26-L5 carcinoma cells by suppressing immune responses in mice Life Sci 66(19) 1827–38 DOI: 10.1016/S0024-3205(00)00506-3 PMID: 10809180
149. Kanemi O et al (2005) Acute stress reduces intraparenchymal lung natural killer cells via beta-adrenergic stimulation Clin Exp Immunol 139(1) 25–34 DOI: 10.1111/j.1365-2249.2005.02672.x
150. Tarr AJ et al (2012) β-Adrenergic receptor mediated increases in activation and function of natural killer cells following repeated social disruption Brain Behav Immun 26(8) 1226–38 DOI: 10.1016/j.bbi.2012.07.002 PMID: 22796551 PMCID: 3468689
151. Ben-Eliyahu S (2012) Can we really know if a stressor increases or decreases natural killer cell activity? Brain Bbehav Immun 26(8) 1224–5 DOI: 10.1016/j.bbi.2012.08.004
152. Sonnenfeld G et al (1992) Stress-induced alterations in interferon production and class II histocompatibility antigen expression Brain Behav Immun 6(2) 170–8 DOI: 10.1016/0889-1591(92)90016-H PMID: 1504370
153. Khalili A et al (2013) Long acting propranolol and HSP-70 rich tumor lysate reduce tumor growth and enhance immune response against fibrosarcoma in Balb/c mice Iran J Immunol 10(2) 70–82 PMID: 23811546
154. Huang H-W et al (2013) Lymphocyte-derived catecholamines induce a shift of Th1/Th2 balance toward Th2 polarization Neuroimmunomodulation 20(1) 1–8 DOI: 10.1159/000343099
155. Huang HW et al (2015) Regulation of differentiation and function of helper T cells by lymphocyte-derived catecholamines via α1- and β2-adrenoceptors Neuroimmunomodulation 22(3) 138–51 DOI: 10.1159/000360579
156. Panina-Bordignon P et al (1997) Beta2-agonists prevent Th1 development by selective inhibition of interleukin 12 J Clin Invest 100(6) 1513–9 DOI: 10.1172/JCI119674 PMID: 9294119 PMCID: 508332
157. Khaled YS, Ammori BJ and Elkord E (2013) Myeloid-derived suppressor cells in cancer: recent progress and prospects Immunol Cell Biol 91(8) 493–502 DOI: 10.1038/icb.2013.29 PMID: 23797066
158. Motallebnezhad M et al (2015) The immunobiology of myeloid-derived suppressor cells in cancer Tumour Biol 37(2) 1387–406 DOI: 10.1007/s13277-015-4477-9 PMID: 26611648
159. Jin J et al (2013) Chronic psychological stress induces the accumulation of myeloid-derived suppressor cells in mice PloS One 8(9) e74497 DOI: 10.1371/journal.pone.0074497 PMID: 24058577 PMCID: 3776856
160. Mao Y et al (2014) Inhibition of tumor-derived prostaglandin-E2 blocks the induction of myeloid-derived suppressor cells and recovers natural killer cell activity Clin Cancer Res 20(15) 4096–106 DOI: 10.1158/1078-0432.CCR-14-0635 PMID: 24907113
161. Zhou L et al (2016) Propranolol Attenuates Surgical Stress-Induced Elevation of the Regulatory T Cell Response in Patients Undergoing Radical Mastectomy J Immunol 196(8) 3460–9 DOI: 10.4049/jimmunol.1501677 PMID: 26969754
162. Normanno N et al (2006) Epidermal growth factor receptor (EGFR) signaling in cancer Gene 366(1) 2–16 DOI: 10.1016/j.gene.2005.10.018
163. Rush JS et al (2012) Endosomal accumulation of the activated epidermal growth factor receptor (EGFR) induces apoptosis J Biol Chem 287(1) 712–22 DOI: 10.1074/jbc.M111.294470 PMCID: 3249126
164. Mosesson Y, Mills GB and Yarden Y (2008) Derailed endocytosis: an emerging feature of cancer Nat Rev Cancer 8(11) 835–50 DOI: 10.1038/nrc2521 PMID: 18948996
165. Shaughnessy R et al (2014) Epidermal growth factor receptor endocytic traffic perturbation by phosphatidate phosphohydrolase inhibition: New strategy against cancer FEBS J 281(9) 2172–89 DOI: 10.1111/febs.12770 PMID: 24597955
166. Norambuena A et al (2010) Phosphatidic acid induces ligand-independent epidermal growth factor receptor endocytic traffic through PDE4 activation Mol Biol Cell 21(16) 2916–29 DOI: 10.1091/mbc.E10-02-0167 PMID: 20554760 PMCID: 2921116
167. Lavie Y, Piterman O and Liscovitch M (1990) Inhibition of phosphatidic acid phosphohydrolase activity by sphingosine. Dual action of sphingosine in diacylglycerol signal termination FEBS Lett 277(1–2) 7–10 DOI: 10.1016/0014-5793(90)80796-L PMID: 2269371
168. Kang F et al (2014) Propranolol inhibits glucose metabolism and 18F-FDG uptake of breast cancer through posttranscriptional downregulation of hexokinase-2 J Nucl Med 55(3) 439–45 DOI: 10.2967/jnumed.113.121327 PMID: 24504055 PMCID: 4564063
169. Nakanishi M and Rosenberg DW (2013) Multifaceted roles of PGE2 in inflammation and cancer Semin Immunopath 35(2) 123–137 DOI: 10.1007/s00281-012-0342-8
170. Nagaraja AS et al (2015) Sustained adrenergic signaling leads to increased metastasis in ovarian cancer via increased PGE2 synthesis Oncogene 35(18) 2390–7 DOI: 10.1038/onc.2015.302 PMID: 26257064 PMCID: 4749473
171. Gay LJ and Felding-Habermann B (2011) Contribution of platelets to tumour metastasis Nat Rev Cancer 11(2) 123–34 DOI: 10.1038/nrc3004 PMID: 21258396
172. Su BB et al (2014) Aspirin may modify tumor microenvironment via antiplatelet effect Med Hypotheses 1 7–9
173. Mikami J et al (2015) Antitumor effect of antiplatelet agents in gastric cancer cells: an in vivo and in vitro study Gastric Cancer 19(3) 817–26 DOI: 10.1007/s10120-015-0556-2 PMID: 26486505
174. Mousa S a and Petersen LJ (2009) Anti-cancer properties of low-molecular-weight heparin: preclinical evidence Thromb Haemost 102(2) 258–67 PMID: 19652876
175. Pfankuchen DB et al (2015) Low molecular weight heparin tinzaparin antagonizes cisplatin resistance of ovarian cancer cells Biochem Pharmacol 97(2) 147–57 DOI: 10.1016/j.bcp.2015.07.013 PMID: 26239805
176. Spano D et al (2013) Dipyridamole prevents triple-negative breast-cancer progression Clin Exp Metastasis 30(1) 47–68 DOI: 10.1007/s10585-012-9506-0
177. Bonten TN et al (2014) Effect of β-blockers on platelet aggregation: a systematic review and meta-analysis Br J Clin Pharmacol 78(5) 940–9 DOI: 10.1111/bcp.12404 PMID: 24730697 PMCID: 4243868
178. Punda A et al (2005) Effects of atenolol and propranolol on platelet aggregation in moderate essential hypertension: randomized crossover trial Croat Med J 46(2) 219–24 PMID: 15849842
179. Aschbacher K et al (2013) Good stress, bad stress and oxidative stress: Insights from anticipatory cortisol reactivity Psychoneuroendocrinology 38(9) 1698–708 DOI: 10.1016/j.psyneuen.2013.02.004 PMID: 23490070 PMCID: 4028159
180. Cao L et al (2010) Environmental and genetic activation of a brain-adipocyte BDNF/leptin axis causes cancer remission and inhibition Cell 142(1) 52–64 DOI: 10.1016/j.cell.2010.05.029 PMID: 20603014 PMCID: 3784009
181. Buehler D et al (2014) Angiosarcoma outcomes and prognostic factors: a 25-year single institution experience Am J Clin Oncol 37(5) 473–9 DOI: 10.1097/COC.0b013e31827e4e7b
182. Penel N et al (2011) Angiosarcoma: state of the art and perspectives Crit Rev Oncol Hematol 80(2) 257–63 DOI: 10.1016/j.critrevonc.2010.10.007
183. Hermans DJJ et al (2011) Kaposiform hemangioendothelioma with Kasabach-Merritt syndrome: a new indication for propranolol treatment J Pediatr Hematol Oncol 33(4) e171-3 DOI: 10.1097/MPH.0b013e3182152e4e PMID: 21516018
184. Ozeki M, Fukao T and Kondo N (2011) Propranolol for intractable diffuse lymphangiomatosis N Engl J Med 364(14) 1380–2 DOI: 10.1056/NEJMc1013217 PMID: 21470038
185. Coghlin C and Murray GI (2010) Current and emerging concepts in tumour metastasis J Pathol 222(1) 1–15 DOI: 10.1002/path.2727 PMID: 20681009
186. Shaked Y (2016) Balancing efficacy of and host immune responses to cancer therapy: the yin and yang effects Nat Rev Clin Oncol 13(10) 611–26 DOI: 10.1038/nrclinonc.2016.57 PMID: 27118493
187. Horowitz M et al (2015) Exploiting the critical perioperative period to improve long-term cancer outcomes Nat Rev Clin Oncol 12(4) 213–26 DOI: 10.1038/nrclinonc.2014.224 PMID: 25601442
188. Antonio N et al (2015) The wound inflammatory response exacerbates growth of pre-neoplastic cells and progression to cancer EMBO J 34(17) 2219–36 DOI: 10.15252/embj.201490147 PMID: 26136213 PMCID: 4585460
189. Neeman E and Ben-Eliyahu S (2013) Surgery and stress promote cancer metastasis: new outlooks on perioperative mediating mechanisms and immune involvement Brain Behav Immun 30 Suppl S32–40 DOI: 10.1016/j.bbi.2012.03.006
190. Gottschalk A et al (2010) The role of the perioperative period in recurrence after cancer surgery Anesth Anal 110(6) 1636–43 DOI: 10.1213/ANE.0b013e3181de0ab6
191. Opitz I et al (2013) Perioperative diclofenac application during video-assisted thoracic surgery pleurodesis modulates early inflammatory and fibrinolytic processes in an experimental model Eur Surg Res 50(1) 14–23 DOI: 10.1159/000341670 PMID: 23429206
192. Shakhar G and Ben-Eliyahu S (2003) Potential prophylactic measures against postoperative immunosuppression: could they reduce recurrence rates in oncological patients? Ann Surg Oncol 10(8) 972–92 DOI: 10.1245/ASO.2003.02.007 PMID: 14527919
193. Horowitz M et al (2015) Exploiting the critical perioperative period to improve long-term cancer outcomes Nat Rev Clin Oncol 12(4) 213–26 DOI: 10.1038/nrclinonc.2014.224 PMID: 25601442
194. Ash S and Buggy DJ (2013) Does regional anaesthesia and analgesia or opioid analgesia influence recurrence after primary cancer surgery? An update of available evidence Best PracRes Clin Anaesthesiol 27(4) 441–56 DOI: 10.1016/j.bpa.2013.10.005
195. Heaney Á and Buggy DJ (2012) Can anaesthetic and analgesic techniques affect cancer recurrence or metastasis? Bri J Anaesth 109(SUPPL1) i17–i28 DOI: 10.1093/bja/aes421
196. Forget P et al (2013) Perioperative ketorolac in high risk breast cancer patients. Rationale, feasibility and methodology of a prospective randomized placebo-controlled trial Med Hypotheses 81(4) 707–12 DOI: 10.1016/j.mehy.2013.07.033 PMID: 23937996
197. Retsky M et al (2012) NSAID analgesic ketorolac used perioperatively may suppress early breast cancer relapse: particular relevance to triple negative subgroup Breast Cancer Res Treat 134(2) 881–8 DOI: 10.1007/s10549-012-2094-5 PMID: 22622810
198. Guo Y et al (2015) A Novel Pharmacologic Activity of Ketorolac for Therapeutic Benefit in Ovarian Cancer Patients Clin Cancer Res 21(22) 5064–72 DOI: 10.1158/1078-0432.CCR-15-0461 PMID: 26071482 PMCID: 4644688
199. Forget P et al (2014) Intraoperative use of ketorolac or diclofenac is associated with improved disease-free survival and overall survival in conservative breast cancer surgery Br J Anaesth 113(Suppl 1) i82–7 DOI: 10.1093/bja/aet464 PMID: 24464611
200. Forget P et al (2013) Neutrophil: lymphocyte ratio and intraoperative use of ketorolac or diclofenac are prognostic factors in different cohorts of patients undergoing breast, lung, and kidney cancer surgery Ann Surg Oncol 20(Suppl 3) S650–60 DOI: 10.1245/s10434-013-3136-x PMID: 23884751
201. Pantziarka P et al (2014) Repurposing drugs in oncology (ReDO)-cimetidine as an anti-cancer agent Ecancermedicalscience 8 485 DOI: 10.3332/ecancer.2014.485 PMID: 25525463 PMCID: 4268104
202. Deva S and Jameson M (2012) Histamine type 2 receptor antagonists as adjuvant treatment for resected colorectal cancer Cochrane Database Syst Rev 8(8) CD007814 PMID: 22895966
203. Eng JW-L et al (2014) A nervous tumor microenvironment: the impact of adrenergic stress on cancer cells, immunosuppression, and immunotherapeutic response Cancer Immun Immunother 63(11) 1115–28 DOI: 10.1007/s00262-014-1617-9
204. Thaker PH, Sood AK and Ramondetta LM (2013) Importance of adrenergic pathways in women’s cancers Cancer Biomarkers 13(3) 145–54
205. Pantziarka P et al (2016) Repurposing Drugs in Oncology (ReDO)-diclofenac as an anti-cancer agent Ecancermedicalscience 10 610 DOI: 10.3332/ecancer.2016.610 PMID: 26823679 PMCID: 4720497
206. Zhang M-F et al (2015) Effectiveness of mindfulness-based therapy for reducing anxiety and depression in patients with cancer: a meta-analysis Medicine (Baltimore) 94(45) e0897–0 DOI: 10.1097/MD.0000000000000897
207. Stagl JM et al (2015) A randomized controlled trial of cognitive-behavioral stress management in breast cancer: survival and recurrence at 11-year follow-up Breast Cancer Research Treat 154(2) 319–28 DOI: 10.1007/s10549-015-3626-6
208. Fancourt D et al (2016) Singing modulates mood, stress, cortisol, cytokine and neuropeptide activity in cancer patients and carers Ecancermedicalscience 10 631 DOI: 10.3332/ecancer.2016.631 PMID: 27170831 PMCID: 4854222




